Retour sur la 13ème journée annuelle du Centre de référence des maladies respiratoires

En raison de la pandémie Covid-19, la 13éme journée annuelle du centre de référence des maladies respiratoires rares (RespiRare) a dû se réorganiser en événement 100 % virtuel. De nombreuses personnes dont des représentants associatifs ont répondu présents à cette matinée du 17 janvier 2022 afin d’échanger autour d’un programme centré sur les actions phares du plan national maladies rares (PNMR 3).
Au programme des 5 groupes de travail RespiRare : les réunions de concertation pluridisciplinaires (RCP) pour le diagnostic et la prise en charge des cas complexes, les principales actualités scientifiques de l’année écoulée et les futurs projets de recherche, la restructuration du réseau dans le cadre de la labellisation des centres maladies rares.
L’année en revue : Malfpulm, PNDS…
Parmi l’actualité phare présentée par le Pr Christophe Delacourt (Hôpital Necker-Enfants malades, AP-HP) figure, la publication en 2021 du protocole de diagnostic et de soins (PNDS) sur les malformations pulmonaires congénitales de l’enfant élaboré par un groupe de travail multidisciplinaire incluant des pneumo-pédiatres, des chirurgiens infantiles, des obstétriciens, des radiologues, des anatomopathologistes, etc. Selon lui, les affirmations fortes révélées dans ce PNDS sont :
- la nécessité (en pratique clinique) de décrire les phénotypes des malformations pulmonaires (et non l’utilisation d’une description histologique) ;
- la place des marqueurs prénataux telle que le CVR (Congenital pulmonary airway malformation Volume Ratio), marqueur prédictif des complications prénatales et néonatales. Il permet de donner aux familles une information nettement améliorée sur les risques ;
- l’histoire naturelle hétérogène mais asymptomatique pour la majorité de enfants ;
- l’indication opératoire pour les malformations kystiques.
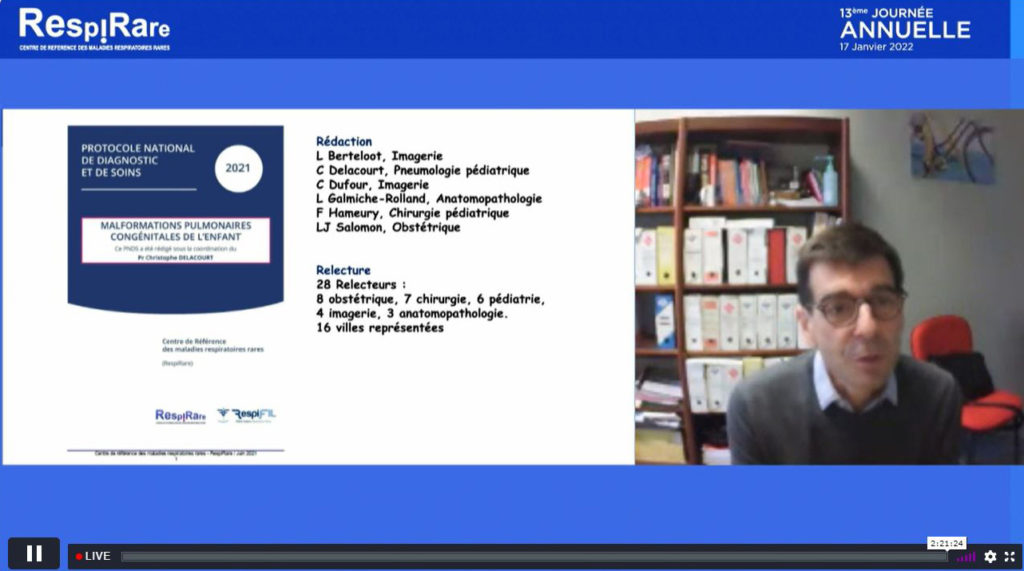

Par ailleurs, le Pr Delacourt rapporte les résultats d’une étude menée par Sergei M. Hermelijn et al. Human pathology (2020), dont l’objectif était d’évaluer le potentiel de divers biomarqueurs immunohistochimiques et génomiques pour prédire la présence de proliférations mucineuses dans les malformations congénitales des voies aériennes pulmonaires (CPAM). En analysant 50 pièces chirurgicales (50 % CPAM de type 1 et 50 % CPAM de type 2), des corrélations imparfaites entre la relecture et l’interprétation du scanner et le compte-rendu histopathologique initial ont été notées. Par ailleurs, la présence de plage de métaplasie mucineuse est rapportée depuis plusieurs années comme étant associée à des mutations somatiques de l’oncogène KRAS. Le séquençage de 23 tissus malformatifs kystiques (12 CPAM de type 1, 11 CPAM de type 2) a mis en évidence de telles mutations dans les proliférations mucineuses et le tissu CPAM non mucineux adjacent, alors que le tissu pulmonaire normal environnant était négatif. En revanche, dans moins de la moitié des échantillons témoins dépourvus de proliférations mucineuses (5 sur 12), le tissu CPAM portait également une mutation KRAS. L’étude de Chang et al. Hystopathology (2021), confirme ces résultats puisque des associations d’adénocarcinomes mucineux et de malformations pulmonaires y compris dans les CPAM de type 2 sont également retrouvées. A partir de la cohorte prospective multicentrique française MALFPULM, des analyses moléculaires sont en cours et feront l’objet d’une prochaine publication.
Bilan des Réunions de Concertation Pluridisciplinaires (RCP) et projets
Les RCP
Le Dr Frédéric Hameury (Hospices Civils de Lyon, HCL), coordonnateur de la RCP nationale ADP est intervenu sur le sujet, en rappelant son organisation, son déroulement et en présentant les membres du quorum (radiologues, pédiatres, chirurgiens pédiatriques, etc.). Depuis mai 2020, 48 dossiers complexes ont été discutés (malformations pulmonaires congénitales, agénésies pulmonaires, hypoplasie, pathologies de la trachée et des bronches, bronchectasies, masse médiastinale, masse pulmonaire etc.) avec des avis diagnostics ou thérapeutiques en anténatal ou post-natal. Pour les avis urgents, il est possible de faire appel au « Comité Thorax ». Du fait de leur récurrence, le 4 mai 2022 (de 16h à 18h), un webinaire sur les agénésies pulmonaires (conséquences et prise en charge) sera organisé à l’initiative de la société française de chirurgie pédiatrique – SFCP.

Les études cliniques en cours

DDB idiopathique de la physiopathologie aux pistes thérapeutiques

Le Dr Rachad El Hajj (centre hospitalier de Versailles), a rappelé un chiffre clé, celui de la prévalence des dilations des bronches (DDB) chez l’adulte, estimée à 1/750 dans les pays occidentaux, dont 80 % rapportent des symptômes respiratoires chroniques dans l’enfance, suggérant un début précoce de cette maladie. Parmi ces DDB, 40 % en moyenne sont idiopathiques, c’est-à-dire sans cause connue ; 60 % peuvent être post-infectieuse, ou liées à d’autres maladies (hors mucoviscidose) telles que les déficiences de l’immunité, les dyskinésies ciliaires primitives (anomalie du fonctionnement des cils bronchiques), etc. De nombreux facteurs peuvent contribuer à la genèse des DDB idiopathiques, notamment les exacerbations inflammatoires répétées, les anomalies structurelles du tissu du soutien ou les anomalies du transport ionique qui peuvent être à l’origine d’un échec de la clairance mucociliaire et augmenter les dilatations idiopathiques des bronches, conduisant, in fine à une insuffisance respiratoire chronique.
L’épithélium pulmonaire, bien que cela soit moins connu, joue un rôle important dans le transport ionique, ce qui contribue à réguler finement la quantité de liquide de surface respiratoire. Ce transport tient à l’activité d’un canal sodium (ENaC) exprimé à la surface apicale des cellules épithéliales. C’est l’absorption du sodium par les cellules épithéliales en conjonction avec la sécrétion des ions de chlorure qui module la réabsorption du liquide des voies aériennes. Les canaux (ATP124, SLC26A4) quant à eux, contribuent à l’homéostasie du pH et à l’acidité du liquide. La modulation fine de ce liquide de surface respiratoire est essentielle au bon déroulement de la clairance mucociliaire, le processus qui permet aux cellules ciliées, en conjonction avec la couche de mucus qui recouvre ce liquide, d’éliminer les micro-organismes pathogènes et les particules qui pénètrent dans les poumons.
Dans l’étude menée à l’hôpital Necker-Enfants Malades (AP-HP) chez 25 enfants atteints de DDB, comparativement à 8 enfants témoins, le Dr Rachad El Hajj rapporte une inflammation plus élevée (en moyenne) dans le liquide du lavage broncho-alvéolaire (LBA) effectué au cours d’une bronchoscopie dans le groupe DDB même si celle-ci n’est pas significative. L’analyse génétique a révélé : trois variants pathogènes pour la sous-unité du canal ENaC, le canal transporteur des ions chlorure (CFTR-RD) ainsi que le canal SLC26A4 ; six variants probablement pathogènes et seize non pathogènes. « L’étude des transporteurs des canaux ioniques confrontée à l’étude génétique est importante pour notamment proposer un traitement symptomatique adaptée ou pour s’orienter vers une médecine personnalisée dans le cas d’une pathologie liée à une mutation CFTR » conclut le spécialiste.
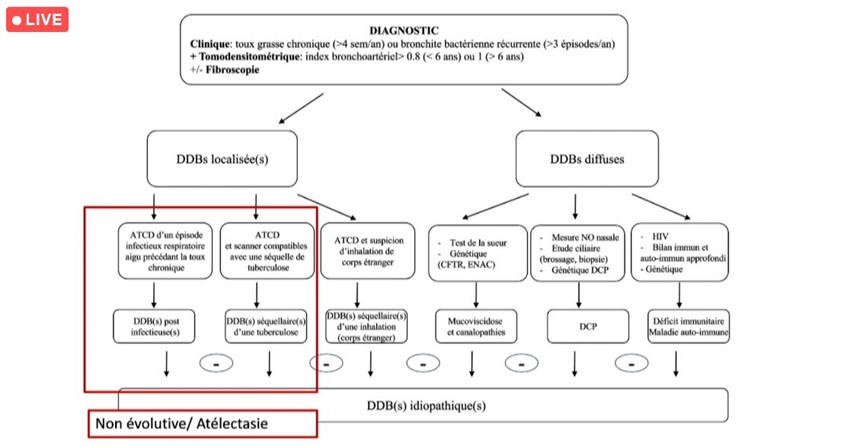
La RCP DDB idiopathique : un instantané des demandes depuis sa création
Depuis fin 2019, la réunion de concertation pluridisciplinaire (RCP) nationale DDB pédiatrique (non CF, non DCP) a permis la discussion de 39 dossiers rapporte le Pr Véronique Houdouin (Hôpital Robert Debré, AP-HP), coordonnatrice de cette RCP. Parmi les diagnostics évoqués lors de ces réunions : la bronchiolite oblitérante (chez 20 % des patients), la bronchopathie chronique post-infectieuse (chez 17 % des patients), les DDB idiopathiques (chez 5,1 % des patients). Chez 20 % des patients, un bilan complémentaire a été demandé pour une pathologie ENaC, une DCP, ou pour un déficit immunitaire (15 %).

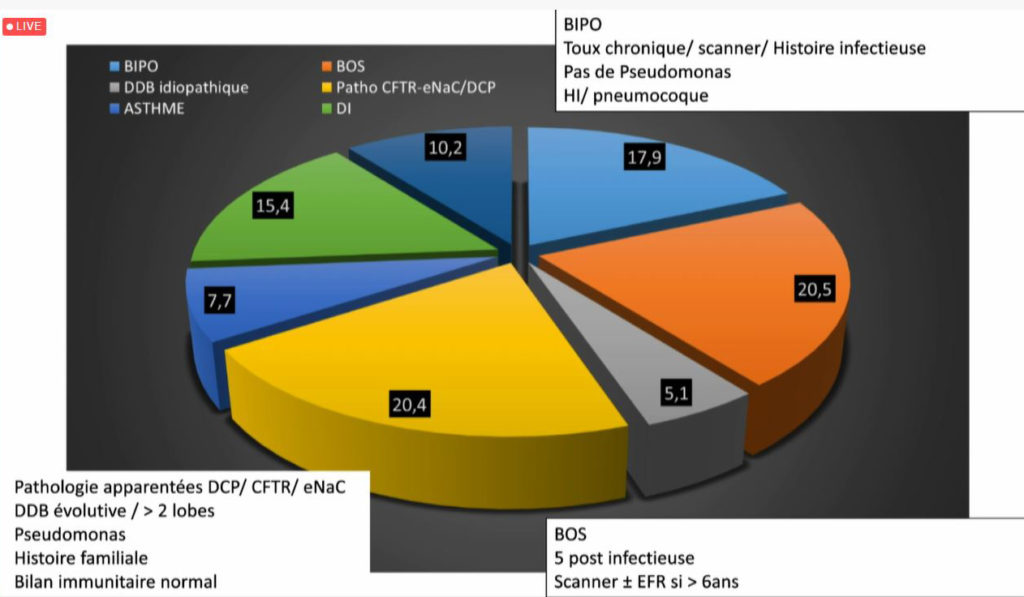
« Lorsque la DDB est localisée, le plus souvent, elle est post-infectieuse ou séquellaire pour laquelle, le diagnostic est assez simple avec une évolution stable (souvent une atélectasie au scanner). Cependant, la recherche de la cause des DDB diffuses est souvent plus complexe. Dans la plupart des cas, des examens complémentaires sont demandés » explique la spécialiste.
La génétique dans le syndrome d’Ondine
Cette session a débuté par la présentation du Dr Nathalie Couque (Hôpital Robert Debré, AP-HP) sur la génétique dans le syndrome d’Ondine. Dans les années 80, les cas intrafamiliaux de syndrome d’Ondine suggèrent une transmission génétique, qui a été établie en 2003 par une équipe française suite à la découverte d’une mutation sur le gène PHOX-2B. Le diagnostic du syndrome d’Ondine est confirmé par l’association d’une hypoventilation alvéolaire ou d’apnées par anomalies du contrôle central de la respiration, avec la présence d’une mutation du gène PHOX-2B.
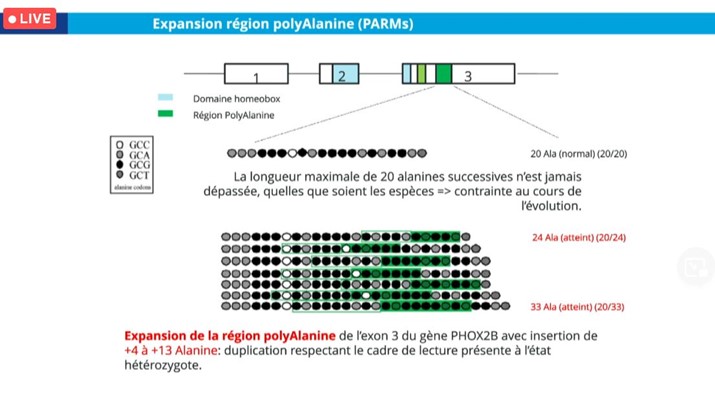
Le gène Paired-like Homeobox 2B (PHOX-2B) code un facteur de transcription qui régit le développement et le fonctionnement du système nerveux autonome (SNA). Il possède un domaine homeobox hautement conservé et deux régions polyalanine. Parmi les mutations décrites dans cette maladie, le Dr Nathalie Couque cite :
- Une expansion de la région polyalanine (= duplication in-frame) dans 90 % des cas aussi appelée : polyalanine repeat mutations (PARMs). Une corrélation génotype/phénotype a été établie entre la longueur de l’expansion et la sévérité clinique. Ainsi, pour les patients présentant un génotype 20/26, la pénétrance est complète et un support ventilatoire est nécessaire mais rarement en continu. Par contre, à partir du génotype 20/27 à 20/33, un support ventilatoire est nécessaire en continu, avec une association avec la maladie de Hirshsprung et plus rarement avec une tumeur de la crête neurale.
- Une mutation ponctuelle tout au long du gène dans 10 % des cas aussi appelée non polyalanine repeat mutations (NPARMs). A ce jour, 130 mutations différentes sont décrites. L’association est significativement plus fréquente entre les NPARMs et la forme sévère syndromique avec un support ventilatoire sur 24h et la maladie de Hirshsprung et/ou le neuroblastome associés. Une minorité des NPARMs présente une pénétrance incomplète avec : une maladie de Hirshsprung isolée ou avec un impact respiratoire limité ; une tumeur de la crête neurale isolée ou avec un impact respiratoire limité ; une hypoventilation centrale isolée avec phénotype variable au sein d’une même fratrie.
- Une délétion complète ou partielle du gène dans < 1 % des cas
S’agissant d’une maladie génétique, elle se transmet selon un mode autosomique dominant : si un des deux parents est porteur du gène défectueux, le risque d’avoir un enfant malade est de 50 % à chaque grossesse. L’analyse génétique des parents est toujours proposée et recommandée. L’absence de la mutation chez les parents permet de confirmer le caractère de novo de la mutation.
La mise en évidence de la mutation ou de mosaïque somatique chez un parent de patient atteint du syndrome d’Ondine a des conséquences sur le conseil génétique (risque de récurrence élevé chez un autre enfant, voisin de 50 %). « Un diagnostic prénatal peut être proposé à tous parents ayant déjà un enfant atteint et caractérisé sur le plan moléculaire (y compris dans le cas de la mutation de novo). A noter que l’analyse d’exome ne permet pas d’éliminer le diagnostic de syndrome d’Ondine. L’ouverture de la pré-indication du PFMG2025 aux hypoventilations centrales non expliquées avec exploration du gène PHOX-2B négative, est une réelle opportunité pour découvrir de nouveaux variants pour de nouveaux patients » conclut le Dr Nathalie Couque.

Bilan et projets du CRMR syndrome d’Ondine
Le Dr Ha Trang (Hôpital Robert Debré, AP-HP) a présenté les principales actions menées par les centres de référence et de compétence : le programme d’éducation thérapeutique du patient (ETP) « Bien vivre et bien respirer avec le syndrome d’Ondine », le bilan de la RCP anomalies du contrôle respiratoire et insuffisances respiratoires chroniques (1 fois/par trimestre) pour l’année 2021, coordonnée par le Dr Jessica Taytard (Hôpital Armand Trousseau, AP-HP), ainsi que les différentes études cliniques en cours.

Le Dr Jessica Taytard a par ailleurs, fait le point sur deux études multicentriques françaises sur le sommeil du nourrisson de 0 à 6 mois. La première porte sur la respiration périodique chez l’enfant âgé de plus de trois mois et né à terme et la deuxième concerne les apnées idiopathiques chez les nourrissons âgés de 0 à 6 mois.
Le Dr Eytan Sarfati (Créteil) introduit la session par une revue des meilleurs articles de l’année 2021 sur les PID.

Exposome inorganique et sarcoïdose pulmonaire pédiatrique : l’étude PEDIASARC (Dr N. Nathan, Paris)
Cette étude observationnelle, rétrospective et multicentrique comporte 36 cas d’enfants ayant une sarcoïdose. Ces enfants ont été appariés à des témoins, sur des critères d’âge et de sexe pour le premier groupe, et avec des enfants atteints de drépanocytose pour le second groupe. Un score d’exposition aux particules inorganiques a ensuite été créé, qui a permis de montrer une différence significative quant à l’exposition indirecte des patients ayant une sarcoïdose pédiatrique. L’exposition indirecte signifie que les co-résidents, majoritairement les parents, ont été exposés dans leur activité professionnelle à des poussières métalliques, des réactifs abrasifs, etc.. En revanche, l’étude n’a pas relevé de différence significative en ce qui concerne l’exposition directe des enfants, ainsi que dans l’exposition hors cadre professionnel des co-résidents. Cette étude était la première à mettre en lien l’exposition environnementale à des particules inorganiques et les sarcoïdoses pédiatriques.
Vasculopathies de l’enfant associées à des mutations du gène STING (Dr M.L. Frémond, Paris)
Cette étude observationnelle, descriptive, rétrospective et multicentrique a décrit 21 patients ayant une vasculopathie associée à des mutations du gène STING.
Les résultats ont montré :
- chez 13 patients sur 21, la mutation se trouvait dans STING1, avec la substitution d’une méthionine par une valine en position 155
- que la symptomatologie débute dans l’enfance autour de 8,5 mois avec des patients qui ont une atteinte pulmonaire (PID, qui peut évoluer vers de la fibrose ; 6 adolescents ont développé une insuffisance respiratoire terminale) et une atteinte cutanée (rash, ulcères, perforation de la cloison nasale, etc.)
- que certains patients avaient besoin d’oxygène, d’autres de ventilation non-invasive
- des opacités en verre dépoli, du « crazy paving », des kystes
- d’autres symptômes fréquents : retard staturo-pondéral, déficit immunitaire avec anomalies des lymphocytes T et des CD8 mémoire avec présence d’autoanticorps
- un bénéfice à traiter ces patients de manière précoce par Ruxolitinib
L’étude révèle une grande hétérogénéité clinique avec des spectres d’atteinte qui peuvent ou non se regrouper. Le diagnostic précoce permet la mise en place rapide d’un traitement et adapté, notamment par anti JAK.
Supplémentation par méthionine d’enfants ayant une protéinose alvéolaire avec mutation du gène MARS (Pr A. Hadchouel, Paris)
Cet essai clinique portait sur l’efficacité, la sécurité et la tolérance de la supplémentation en méthionine des patients ayant une protéinose MARS. La supplémentation en méthionine a permis une amélioration sur le plan respiratoire, hépatique et sur la croissance staturo-pondérale. Les scanners des 4 patients traités par méthionine montrent une diminution voire une totale disparition des lésions en verre dépoli et l’aspect de « crazy paving ». Sont également constatés une clairance du matériel lypoprotéique, du matériel extra-cellulaire, permettant un arrêt des thérapeutiques.
En ce qui concerne la tolérance et la sécurité, certains patients ont eu des nausées et vomissements en début de traitement et certains ont eu une légère élévation des transaminases, probablement causés par un sous-dosage de la méthionine. Cette étude a donc montré que la supplémentation en méthionine est associée à des améliorations importantes chez les patients atteints de protéinose alvéolaire liée à une mutation du gène MARS 1. Elle ouvre la voie à des stratégies similaires pour d’autres déficits en ARNt synthétases.
Effet in vitro de la Cyclosporine sur la maturation et la localisation de la protéine ABCA3 (M. Forstner, Allemagne)
Cette étude fondamentale a pour objectif de mettre en évidence un correcteur de ABCA3 dans un modèle cellulaire. Un criblage haut-débit avec plus de 1200 molécules a été réalisé et a permis de retenir la cyclosporine comme étant un correcteur potentiel de maturation pour les mutations de maturation de la protéine ABCA3 présente dans le surfactant. Le modèle cellulaire A549 a permis de mettre en évidence une restauration de la localisation intra-vésiculaire d’ABCA3 lorsque les cellules étaient cultivées avec de la cyclosporine. Une augmentation de la forme mature d’ABCA3 en présence de cyclosporine a également été retrouvée.
L’effet des glucocorticoïdes dans les mutations du gène SFTPC du surfactant sur un modèle ex vivo/in vitro (Dr C. Delestrain, Créteil)
Cette autre étude fondamentale a permis d’analyser le mécanisme d’action des glucocorticoïdes chez les patients ayant une mutation SFTPC. Cela repose sur l’analyse du lavage bronchoalvéolaire (LBA) de patients avant et après la prise de glucocorticoïdes versus des patients témoins. Après 5 mois de traitement par glucocorticoïdes, une amélioration sur le plan clinique est observée, qui se traduit également par une amélioration sur le scanner de contrôle, et l’apparition dans le LBA d’une forme intermédiaire de la pro-protéine pro-SPC. Cela montre que la maturation peut se poursuivre après le traitement. Il a également été constaté une diminution de l’expression totale de l’ARN messager SFTPC.
Transition dans les PID : les programmes de transition entre l’âge pédiatrique et l’âge adulte (V. Koucký, République Tchèque)
Cette étude observationnelle descriptive avait pour objectif de décrire les disparités d’approches de la transition enfant / adulte, des patients atteints de PID. Un questionnaire a été envoyé a une centaine de centres spécialisés dans le monde. Les 39 réponses, venant de 21 pays majoritairement européens, ont permis de montrer que le système actuel de transition est très variable et peu standardisé dans la plupart des centres (absence de spécialistes PID, de radiologues, de procédures établies). Ainsi, moins de 10 centres participent à un registre européen des PID (chILD-EU) et d’autres centres utilisent un registre national. Les auteurs concluent en proposant un protocole standard de transition et un rapport médical de transition.
Tolérance du nintédanib chez les enfants de plus de 6 ans avec une fibrose pulmonaire débutante (R. Deterding, Etats-Unis)
Un essai clinique multicentrique (24 pays 70 sites) prospectif, en double aveugle, randomisé et contrôlé par placebo est en cours et a pour objectif d’inclure 30 enfants avec une fibrose pulmonaire débutante. Il s’agit du 1er essai contrôlé randomisé d’un agent antifibrotique chez des enfants ayant une PID. L’étude se décompose en deux temps : dans un premier temps, 20 patients vont recevoir le nintédanib et seront comparés à 10 patients « placebos » sur une durée de 24 semaines. Ensuite, la 2ème phase inclura davantage de patients. Les principaux critères de jugement sont la pharmacocinétique et les effets indésirables. Les critères secondaires sont l’évolution de la fonction respiratoire, le test de marche et le questionnaire de qualité de vie. L’investigateur principal pour la France est le Pr Ralph Epaud (Créteil), l’âge d’inclusion minimum est de 12 ans.
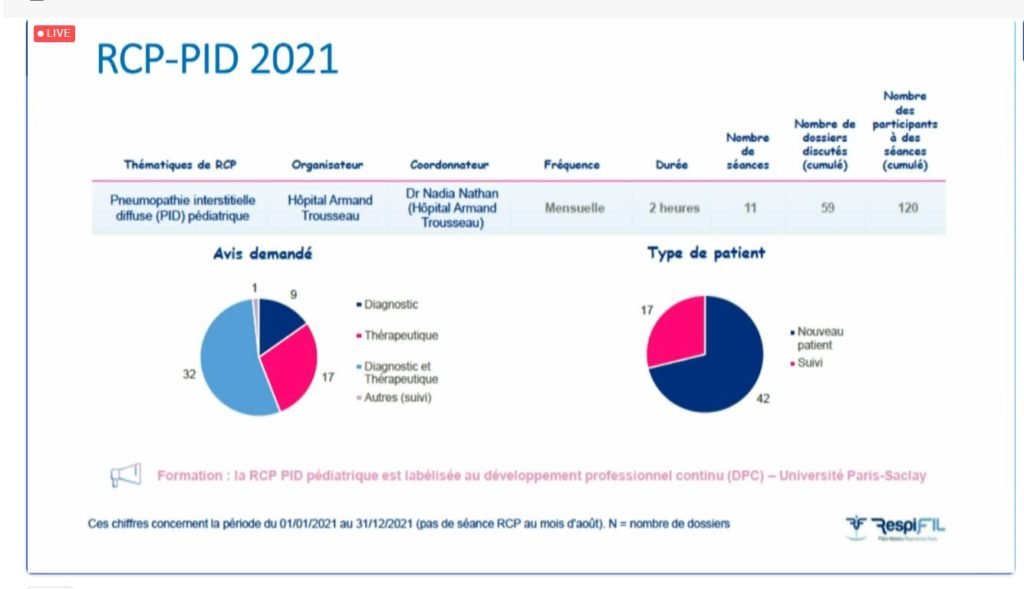
Le Dr Nadia Nathan (Paris) a poursuivi la matinée en présentant les actualités du groupe PID, en commençant par les Réunions de Concertation Pluridisciplinaires (RCP). En 2021, 11 séances ont eu lieu et 59 dossiers ont pu être discutés avec un objectif diagnostic et/ou thérapeutique. La RCP a rassemblé plus de 120 participants sur l’ensemble des séances.
En 2021, de nombreux documents et projets ont vu le jour :
- Des PNDS : le PNDS sur les pathologies du surfactant est désormais disponible sur le site de la Haute Autorité de Santé (HAS). Les PNDS sur la sarcoïdose, l’hyperplasie des cellules neuroendocrines (NEHI), et la dysplasie bronchopulmonaire sont en préparation.
- Des livrets d’information : production d’un livret sur les tests génétiques et traduction en anglais des livrets sur le surfactant et le NEHI pour être implémentés sur le site du groupe européen. Un livret sur la nutrition des enfants atteints de PID un livret sur l’hémosidérose sont en préparation.
- Développement des liens avec les associations de patients : l’AFPIE va fêter ses 10 ans, l’association « Ensemble pour Pedro ». Une nouvelle association est en train d’être créée pour les NEHI.
- 2 programmes d’éducation thérapeutique financés par l’AAP DGOS 2020 : un projet concernant les PID à l’Hôpital Trousseau AP-HP et un projet concernant les PID et les dysplasies bronchopulmonaires au CHI Créteil
Les études cliniques sont également nombreuses :
- Etude nintédanib versus placebo (Boehringer Ingelheim) : 2 enfants ont pu être inclus en France au CHI Créteil. Les inclusions sont terminées pour la première phase (voir étude ci-dessus).
- Etude sur la méthionine dans les protéinoses alvéolaires, coordonnée par la Pr Hadchouel (Paris) est toujours en cours
- Une étude de la variabilité du diagnostic des PID entre les praticiens pédiatriques et adultes va débuter, supervisée par le Pr Ralph Epaud (Créteil)
- Etude sur l’utilisation de l’échographie thoracique pour le diagnostic et le suivi des PID pédiatriques menée par Michaël Schum et Céline Delestrain (Créteil)

Plusieurs études de cohortes sont en cours ou vont débuter prochainement :
| En cours | À venir |
| NEHI à Marseille (Candice Fabre et Jean-Christophe Dubus) | Etude globale d’épidémiologie des PID (Nadia Nathan) |
| SP-C de début pédiatrique au Créteil (Eytan Sarfati, Céline Delestrain) | ABCA3 de début pédiatrique (Camille Fletcher, Nadia Nathan) |
| Maladies auto-immunes sans mutation COPA ni STING, effet des anti-jak sur le poumon à Paris (Alice Hadchouel) | Atteinte respiratoire des mucopolysaccharidoses à Paris (Maxime Le Fevre) |
| Qualité de vie des parents d’enfants atteints de PID à Trousseau (Nadia Nathan) | TBX4 pédiatrique à Créteil (Ralph Epaud) |
Les projets scientifiques en cours :
- L’équipe GEIC20 du laboratoire des interactions génétique-environnement dans la BPCO, la mucoviscidose et autres (rares) pathologies respiratoires de l’hôpital Henri Mondor (Créteil) mène des études structurales et fonctionnelles des mutations du transporteur alvéolaire ABCA3, responsable de pathologies du surfactant. Une étude menée par Eytan Sarfati est également en cours sur le potentiel thérapeutique de petites molécules dans les pathologies du surfactant associées à des mutations d’ABCA3.
- Au laboratoire des maladies génétiques d’expression pédiatrique de Trousseau (Paris), 3 études sont en cours : 1/ Conséquences cellulaires des mutations SFTPA1/SFTPA2 (Yohan Soreze) ; 2/ Etude fonctionnelle des mutations SFTPB et ABCA3 (Tifenn Desroziers) ; 3/ Fibrose pulmonaire et nécroptose (Camille Fletcher)
- Le laboratoire de Necker mène une étude sur les conséquences cellulaires des mutations des ARS sur des fibroblastes de peau.
Au niveau européen également, beaucoup de choses se mettent en place pour les PID :
- Une étude de cohorte sur les hémorragies alvéolaires diffuses menée par Astrid Ring au Danemark. Plus de 100 patients, dont 18 français, sont inclus dans cette cohorte qui va être publiée prochainement.
- Un groupe de l’ERN-LUNG sur les PID pédiatriques, Hôpital Trousseau AP-HP en est un « core member »
- La COST-ENTeR chILD s’est achevée en juillet 2021 mais se poursuit par le « Cost Innovative Grant », avec un volet plus scientifique.
- Le groupe de la Clinical Research Collaboration de l’European Respiratory Society (ERS) : ERS CRC ChILD. Les responsables actuels sont Steve Cunningham (GB) et Nico Schwerk (Allemagne), qui vont être remplacés en mars 2022 par Matthias Griese (Allemagne) et Nadia Nathan (France). Le groupe propose des projets de recherche clinique, des réunions biannuelles et favorise l’inclusion de jeunes participants.
- Une étude sur la transition acceptée comme Task force de l’ERS coordonnée par Petr Pohunek (partie pédiatrique) et Effrosyne Manali (partie adulte).
En 2022, le Dr Nadia Nathan souhaite mettre en place des webinaires réguliers pour permettre la présentation de travaux cliniques ou scientifiques (mémoires, articles), proposer des cas cliniques didactiques. Cela permettrait également de réfléchir à des projets communs, à la mise en place de travaux de recherche ou de Programmes Hospitaliers de Recherche Clinique (PHRC).
Le Pr Philippe Reix (Lyon) entame la session par une revue de la bibliographie. L’activité dans le domaine de la DCP assez dense, quelques articles ont ainsi été sélectionnés.
Comparaison du microbiote des voies aériennes des patients atteints de maladies respiratoires bronchiques suppuratives (B. Ahmed, Royaume-Uni)
L’étude publiée par Ahmed et al. compare le microbiote des voies aériennes des enfants atteints de mucoviscidose et de maladies ciliaires. La cohorte, composée d’un groupe de 31 enfants atteints de DCP et d’un groupe de 31 enfants atteints de mucoviscidose, comparables, a été suivie pendant 1 année. L’étude a révélé de grandes différences en espèce, en diversité et en richesse dans les 2 microbiotes. Parmi les faits marquants, on note beaucoup moins de patients avec Pseudomonas chez les patients DCP par rapport aux patients atteints de mucoviscidose. En revanche, on retrouve beaucoup plus de pneumocoques et d’Haemophilus chez les patients DCP. Dans les 2 groupes, le streptocoque est le germe le plus retrouvé. Le microbiote DCP est moins riche mais plus diversifié que celui des patients atteints de mucoviscidose. En conclusion, le microbiote est très différent dans les 2 groupes. Ainsi, la prise en charge anti-infectieuse des patients atteints de DCP n’est pas toujours adaptée si elle est extrapolée de celle de la mucoviscidose.
Les atélectasies partielles adjacentes à la scissure chez les patients atteints de DCP (L. Fabri, Australie)
Le Pr Philippe Reix présente un second article paru récemment qui pose la question de l’existence d’anomalie radiologique spécifique de la DCP. En effet, une atélectasie partielle adjacente à la scissure (APAS) serait plus souvent retrouvée au cours des DCP et pourrait orienter le diagnostic chez un patient avec un tableau compatible. Les auteurs ont revu 558 scanners de 40 patients enfants et adultes, dont 40 % avaient un situs inversus et 27 % étaient âgés de moins de 18 ans. Une APAS est retrouvée sur 32,5 % des 40 scanners initiaux et chez 29 % des 2-18 ans. Le plus souvent, cette atélectasie était retrouvée au niveau du lobe moyen (84 % des cas). On ne retrouve aucun lien avec le fait d’avoir un situs inversus ou d’avoir une microbiologie particulière. Les auteurs concluent qu’il s’agit d’un signe radiologique intéressant et qui peut être un élément d’orientation pour le diagnostic de DCP, en complément des autres éléments cliniques et paracliniques.
Mesure du NO nasal dans les DCP : attention à la confusion entre DCP et déficit immunitaire primitif (A. Barber, Etats-Unis)
Une équipe américaine a publié un cas clinique sur les limites des mesures de débit du NO nasal. La mesure du débit de NO nasal est en effet une étape importante dans l’algorithme diagnostic des DCP. Pour le débit NO nasal en chimioluminescence, on considère qu’une mesure inférieure à 77 nL/min est très en faveur d’un diagnostic de DCP et incite à pousser plus loin les explorations. Il est par ailleurs connu que certaines pathologies sont à l’origine d’une diminution du débit du NO nasal (obstruction nasale, mucoviscidose, une infection des voies aériennes supérieures…). L’article présente le cas d’une jeune fille, vue pour la 1ère fois à 11 ans avec des antécédents d’infections ORL multiples, et une mesure du VNO basse et une analyse ultrastructurelle normale. L’analyse génétique des 42 gènes DCP n’a retrouvé aucun variant pathologique, la patiente a donc été classée « DCP probable ». Au cours d’une infection à mycobactéries atypiques et un traitement prolongé par antibiotique, la mesure du NO nasal augmente. A 21 ans, la patiente développe une maladie hématologique, suite à laquelle un variant pathologique GATA2 a été retrouvé. Les auteurs illustrent les difficultés diagnostiques des DCP et la nécessité de s’orienter vers un déficit immunitaire primitif si le VNO est bas de façon répétée chez un patient « DCP probable ».
Les recommandations nord-américaines ont par ailleurs été revues et mises à jour en 2020. Elles rappellent qu’une mesure du VNO supérieure à 77 nL/min n’exclut pas le diagnostic de DCP. Certains patients porteurs de mutations de certains gènes sont connus pour avoir une mesure supérieure à cette valeur. Cela représente néanmoins moins de 5 % des patients atteints de DCP. Le point de départ reste identique : il est nécessaire d’avoir une symptomatologie évocatrice, le NO reste à la place des explorations de ces patients. En fonction de la progression dans les examens complémentaires et des résultats, notamment dans les cas pour lesquels il n’y a pas d’analyse ultrastructurale évocatrice ou pas de mutation pathogénique bi-allélique, il faut rechercher et explorer sur le plan génétique la possibilité de certains déficits immunitaires.
Modélisation de la DCP à l’aide d’organoïdes (J. van der Vaart, Pays-Bas)
Une équipe néerlandaise, associée à des chercheurs israéliens, a mis au point les premiers organoïdes réalisés à partir de patients atteints de DCP. Les organoïdes sont des mini-organes (structures cellulaires en 3 dimensions) qui reproduisent par exemple l’épithélium respiratoire ou intestinal. L’équipe est partie de 6 sujets, 2 sains et 4 porteurs de DCP avec des anomalies génétiques identifiées. A partir de prélèvements de brossage nasal, des cultures ont été réalisés dans différents milieux nutritifs. En conditions optimisées, avec du milieu « cilia medium », un plus grand nombre d’organoïdes et surtout une plus grande proportion de cellules ciliées ont été obtenus. Bien que des différences aient été observées entre les différents génotypes, les chercheurs ont retrouvé des caractéristiques compatibles avec le phénotype de départ. Cet outil cellulaire pourrait être optimisé et devenir ainsi dans un futur proche un outil diagnostic, mais également de thérapeutiques pour cibler un certain nombre de molécules sur ces modèles in vitro.
Pour conclure sa présentation, le Pr Philippe Reix mentionne le projet Sinclair, qui vise à comprendre l’évolution de l’index de clairance pulmonaire dans une population pédiatrique de patients atteints de DCP. Il repose sur le recueil de données « en soins courants » dans les centres disposant d’un appareil de LCI. Plusieurs centres ont déjà répondu positivement, le projet débutera suite à la proposition d’un protocole de suivi simple.
Les Réunions de Concertation Pluridisciplinaires DCP
Les RCP DCP fonctionnent bien, ainsi le nombre et la fréquence des séances ont été augmentés. On observe par ailleurs plus de suivi sur la durée des patients présentés en RCP.
Les cartes d’urgence DCP
Bien que les cartes d’urgence DCP soient disponibles depuis plusieurs mois l’association ADCP a remonté des difficultés pour l’obtenir dans certains centres. Pour rappel, ces cartes d’urgence sont disponibles sur demande à l’adresse .
Le film d’information sur la DCP
Plusieurs actions ont été menées par l’association ADCP, avec pour objectif de donner de l’information sur la maladie. Un film d’information sur la DCP à destination des médecins traitants est sorti l’été dernier et un flyer est en train d’être créé avec une aide médicale.
Les programmes d’Education Thérapeutique du Patient (ETP)
Par ailleurs, 2 programmes d’éducation thérapeutique du patient ont été financés par l’appel à projets
DGOS 2019, avec notamment pour objectif de favoriser l’eETP. Le premier projet porté par le Dr Guillaume Thouvenin (Paris) est à destination des enfants d’âge scolaire « Pediacil », le second projet porté par le Dr Natascha Rémus (Créteil) est destiné aux adolescents sur la thématique de la transition. Les équipes, qui travaillent conjointement, ont pu produire un référentiel de compétences, assez commun aux 2 programmes, et déclarer les programmes à l’Agence Régionale de Santé (ARS) en novembre 2021.
Les patients ont été fortement impliqués dans la conception des programmes, notamment via la réalisation de vidéos. L’ambition est de pouvoir proposer ces programmes en ligne sur la plateforme Stimulab pour enrichir les contenus, et pouvoir ainsi proposer plusieurs formats : présentiel, mixte ou e-learning, selon les besoins et envies du patient.

En parallèle, un projet de programme d’Activité Physique Adaptée (APA) est en cours d’élaboration avec la plateforme, afin d’avoir un programme coordonné et national d’activité physique pour les patients atteints de DCP en distanciel.

La recherche
Au niveau de la recherche, en plus du projet Sinclair coordonné par le Pr Philippe Reix (voir ci-dessus), le programme RaDiCo DCP est toujours en cours, coordonné par le Pr Bernard Maitre (successeur du Dr Estelle Escudier). Les premières inclusions ont été faites en 2017, avec une période d’inclusion de 5 ans, avec pour objectif l’identification de critères spécifiques de sévérité et la recherche de corrélation génotype-phénotype. A ce jour, 234 patients ont été inclus et l’objectif est d’inclure 300 patients d’ici le mois de mai 2022.
Les collaborations européennes
Les collaborations européennes autour de la DCP sont également nombreuses avec la création d’un réseau de collaboration européenne au sein de la BEAT-PCD. Bien que le financement de la COST ce soit terminé le projet a pu être poursuivi grâce la création d’un CRC via l’European Respiratory Society (ERS). Les médecins français sont particulièrement impliqués dans les projets suivants :
- La collecte de données standardisées cliniques. Un questionnaire a été créé de manière collaborative et traduit en français. Ces questionnaires pourront être utilisés pour l’ensemble des études menées pour les patients atteints de DCP et permettent de répondre au problème de comparaison de données au niveau européen.
- Le projet EPIC-PCD vient de débuter et est orienté sur les voies aériennes supérieures (réseau d’ORL). Il va permettre de mieux comprendre les signes spécifiques de la maladie, de rechercher des corrélations entre la pathologie ORL et pulmonaire, de décrire l’évolution de la pathologie ORL et d’identifier les critères pronostiques.
- Dans le cadre de l’ERS, la création d’un Clinical Trial Network a débuté. Il a pour objectif de mener des études sur les thérapies d’intérêt dans le cadre de la DCP au niveau international.
- Dans un autre groupe de travail, le Dr Marie Legendre (Paris) travaille sur une base de données européennes pour obtenir des données détaillées sur les gènes et les mutations des gènes impliqués dans les DCP.
Plans Nationaux Maladies Rares & les critères de labellisation des centres experts
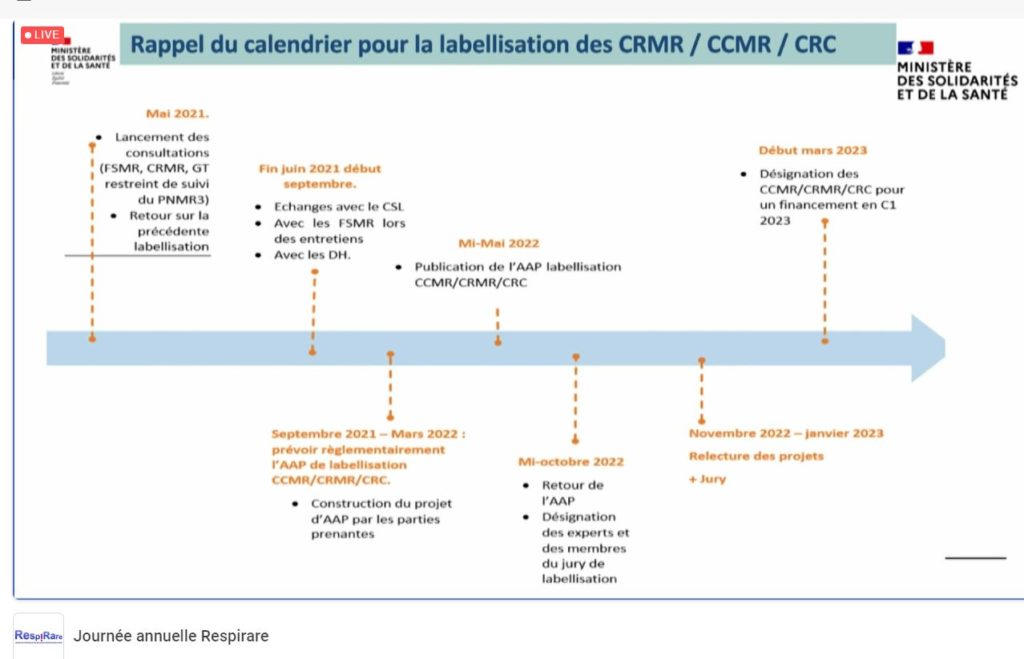
Anne-Sophie Lapointe, cheffe de projet de la Mission Maladies Rares (DGOS), était invitée pour évoquer le Plan National Maladies Rares (PNMR) ainsi que les critères de la labellisation des centres experts à venir. Elle a ainsi pu rappeler que nous en sommes au ¾ du PNMR3, qu’il y a encore beaucoup de projets à finir, notamment les AAP DGOS 2019 et 2020 PNDS, ETP, observatoire du diagnostic, du traitement, et que l’étape préalable à la réflexion d’un PNMR4, sera de dresser un bilan de ce 3ème Plan de façon collégiale en regardant précisément les indicateurs fixés. Elle évoque également que, pour elle, la création récente des plateformes d’expertise maladies rares pourrait permettre de travailleur sur la territorialité dans un futur plan. Dans un 3ème temps, Mme Lapointe a pu préciser le calendrier de la future labellisation : instruction en mai 2022, candidatures à déposer en octobre 2022, jury et labellisation à l’automne pour un premier financement début 2023. Différents critères administratifs ont été détaillés : file active, recherche, formation, etc..
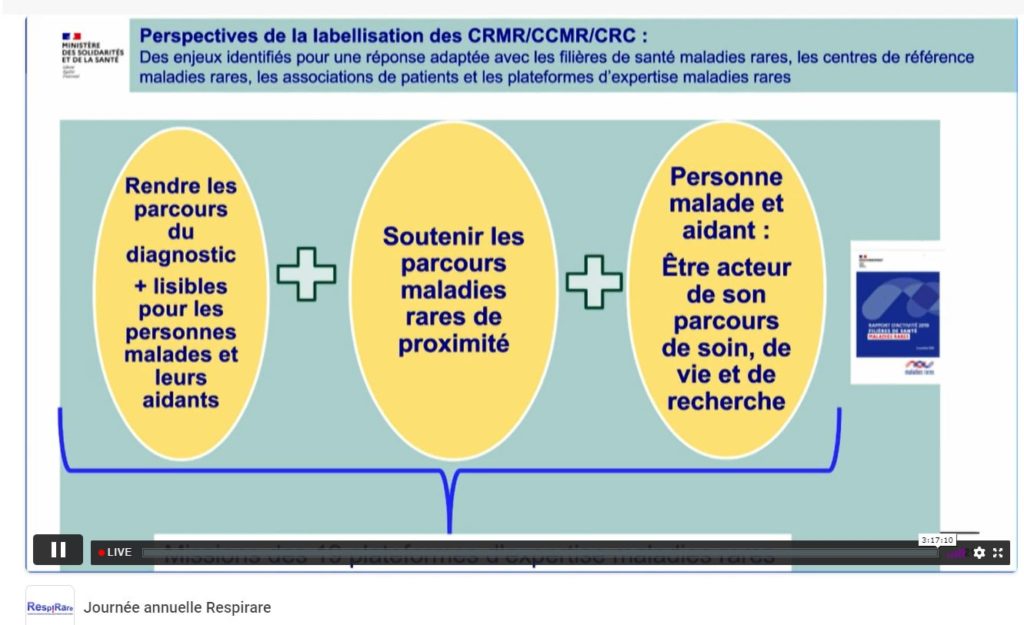
La future labellisation du centre de référence des maladies respiratoire rares (RespiRare)
Durant cette matinée, dans le cadre de la future labellisation des centres de référence et de compétence, la Pr Annick Clement (Hôpital Trousseau AP-HP) a abordé sa succession à la tête du réseau qui reviendrait au Pr Christophe Delacourt (Hôpital Necker Enfants-malades AP-HP).
C’est ainsi que le Pr Christophe Delacourt a pu énoncer les futures orientations du centre de référence des maladies respiratoires rares RespiRare :
- Renforcement du caractère collectif en privilégiant l’alternance de la gouvernance
- Décentralisation des Centres de Référence Maladies Rares (CRMR) constitutifs en province
- Elargissement des Réunions de Concertation Pluridisciplinaires (RCP) aux pays francophones
- Intensification des parcours de formation
- Promotion des métiers
- Optimisation des ressources de financement
- Implication renforcée dans les réseaux européens de référence (ERN-Lung)
- Renforcement de la visibilité des actions RespiRare à l’international
Pour conclure cette matinée, la Pr Annick Clement a remercié toute la communauté RespiRare pour son implication et a souligné que :
Ce fût un honneur de créer RepiRare, sa poursuite n’en est qu’à ses débuts. Tous mes vœux pour 2022 !