Présentation de la maladie
En résumé
Le nom de LymphAngioléioMyomatose reflète les différentes composantes de la maladie :
- Lymphangio se rapporte aux vaisseaux lymphatiques
- Atose réfère au caractère diffus de la maladie
Lymphangioléiomyomatose = maladie diffuse des cellules musculaires lisses de la paroi des vaisseaux lymphatiques
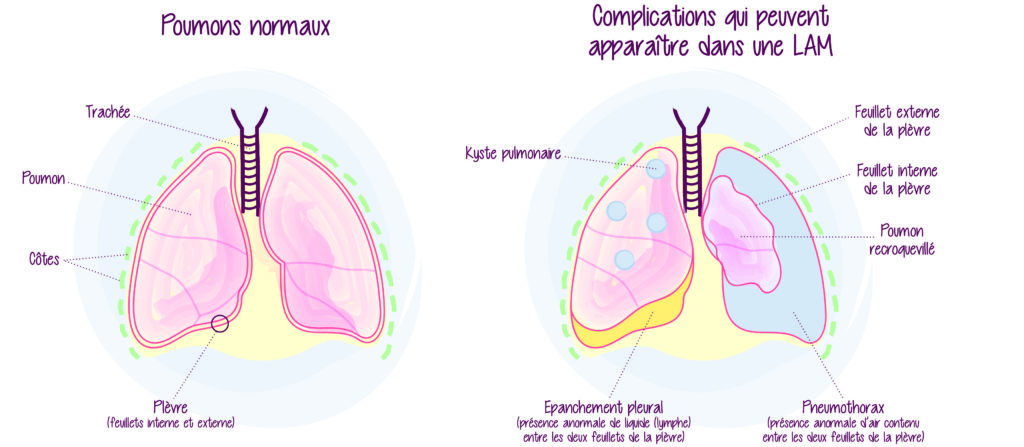
La lymphangioléiomyomatose (ou LAM) est une maladie rare caractérisée par une multiplication excessive des cellules de type musculaire lisse des parois vasculaires, en particulier des vaisseaux lymphatiques de l’organisme et plus spécifiquement des poumons. Les cellules LAM s’accumulent autour des voies respiratoires entraînant la formation de kystes pulmonaires (bulles d’air).
La LAM survient de manière sporadique ou bien associée à une autre maladie rare appelée la sclérose tubéreuse de Bourneville (STB). La STB est une maladie génétique caractérisée par le développement de tumeurs bénignes qui peuvent toucher différents organes, principalement la peau, le cerveau, les yeux, les reins et le cœur.
La LAM sporadique touche principalement les femmes jeunes, en âge de procréer. Il s’agit d’une maladie qui évolue progressivement, entraînant une aggravation progressive des symptômes, le plus souvent respiratoire (essoufflement, toux, douleurs thoraciques), mais qui peuvent être contrôlés par des médicaments.
Personnes concernées
Selon les estimations, trois à cinq femmes sur un million développent une LAM sporadique. Les femmes de 20 à 69 ans représentent la population la plus touchée – l’âge moyen de survenue de cette maladie est de 35 ans. Dans 10 % des cas, la maladie est découverte de façon tardive, après la ménopause.
Causes et risques
La LAM est due à des mutations des gènes tuberous sclerosis complex 1 (TSC1) ou tuberous sclerosis complex 2 (TSC2), qui les rend inactifs. Or, ces deux gènes empêchent l’action d’un facteur de croissance appelé mTOR qui favorise la prolifération et la survie des cellules musculaires lisses. Ainsi, quand l’un des gènes TSC est inactivé, la protéine mTOR reste constamment activée, participant ainsi à la multiplication anormale des cellules musculaires lisses.
Dans la LAM sporadique, la mutation génétique n’existe que dans les cellules musculaires lisses et pas dans les autres cellules, notamment les gamètes qui servent à la reproduction. Cette forme sporadique n’est donc pas héréditaire et n’est pas transmise aux enfants.
En revanche, dans la LAM associée à la STB, les mutations sont héréditaires et touchent toutes les cellules, notamment celles destinées à la reproduction. La STB est une maladie héréditaire dominante c’est-à-dire que le parent atteint de la maladie a 50 % de risque de la transmettre à son enfant.
Symptômes
Les symptômes varient, mais les deux plus fréquents sont l’essoufflement et les douleurs thoraciques témoins d’un pneumothorax (affaissement/décollement du poumon). Le pneumothorax révèle la maladie dans la moitié des cas et récidive chez environ un tiers de ces cas. Il se produit lors de la rupture des kystes pulmonaires remplis d’air, entraînant le passage de l’air dans la cavité pleurale. La cavité pleurale est l’espace entre les poumons et la paroi thoracique.
Parmi les symptômes, les moins fréquents figurent la toux, les tumeurs bénignes des reins (angiomyolipomes) et le chylothorax (accumulation de lymphe dans la cavité pleurale).
Diagnostic
La LAM se manifeste le plus souvent par des difficultés respiratoires qui sont similaires à celles d’autres maladies pulmonaires plus fréquentes. C’est pourquoi, il peut s’écouler plusieurs années avant qu’elle ne soit diagnostiquée. La détection de la LAM repose sur la réalisation d’un scanner (tomodensitométrie ou TDM) thoracique. Ce dernier permet de mettre en évidence les kystes pulmonaires. D’autres signes de la maladie peuvent être mises en évidence comme un pneumothorax, un épanchement pleural liquidien (chylothorax) ou sur une imagerie abdominale des angiomyolipomes du rein. Un test sanguin révélant un niveau élevé d’une protéine appelée facteur de croissance endothélial vasculaire D (VEGF-D) dans le sang peut également permettre de diagnostiquer la LAM.
Dans certains cas, une biopsie pulmonaire peut être nécessaire pour confirmer le diagnostic. Plusieurs méthodes sont possibles comme une biopsie transbronchique ou une cryobiopsie réalisées au cours d’une fibroscopie bronchique ou par une chirurgie (biopsie pulmonaire chirurgicale).
Prise en charge
Bien que l’on ne sache pas la guérir, des progrès significatifs ont été accomplis ces dernières années permettant la prise en charge de la LAM, avec l’objectif d’améliorer les symptômes, la qualité de vie et freiner l’évolution de la maladie.
- Pour limiter l’essoufflement, des bronchodilatateurs inhalés apportent un bénéfice dans un cas sur quatre environ.
- Dans certains cas, le sirolimus® ou rapamycine (habituellement utilisé comme immunosuppresseur dans la transplantation d’organe) peut être utilisé afin de stabiliser, voire améliorer la fonction respiratoire. Il réduit la taille des angiomyolipomes et la récurrence des épanchements de liquides.
- Le pneumothorax est une urgence médicale. Il peut être traité par drainage pour évacuer l’air de la cavité pleurale ou par chirurgie notamment pour éviter les récidives.
- Le traitement du chylothorax comprend principalement des mesures diététiques adaptées (régime pauvre en graisses) et si besoin une ponction pour évacuer l’excès de lymphe.
- Les angiomyolipomes rénaux ne nécessitent aucun traitement s’ils sont de petites tailles. En revanche, s’ils grossissent et/ou entraînent des douleurs ou des saignements, une embolisation (obstruction du vaisseau qui saigne) ou une intervention chirurgicale peuvent être nécessaires.
- Dans de très rares cas, une transplantation pulmonaire peut être proposée dans le cas d’une insuffisance respiratoire très invalidante. Ce traitement est discuté au cas par cas en fonction de l’âge et s’il n’y a pas de contre-indication.
Mesures associées
Il est important que les patientes suivent des programmes d’éducation thérapeutique pour apprendre à gérer leur maladie et reconnaître les signes d’alarme qui nécessitent une consultation. En cas de :
- Contraception : les contraceptifs oraux (pilule) contenant des œstrogènes ont été suspectés pour aggraver la maladie. Il est conseillé d’éviter tous les traitements contenant des œstrogènes, y compris la pilule contraceptive combinée orale (contenant à la fois des estrogènes et de la progestérone) et le traitement hormonal de substitution (dans le cas de la ménopause). En revanche, la pilule microprogestative est autorisée.
- Grossesse : avant d’envisager une grossesse, il est essentiel d’en discuter avec l’équipe en charge du suivi de la LAM, du fait du risque d’aggravation de la maladie. Il faudra donc évaluer ce risque qui est très variable d’une femme à l’autre. Un conseil génétique est proposé aux patientes atteintes de STB avant la grossesse.
- Ostéoporose : une diminution de la densité des os est souvent associée à la LAM. L’ostéoporose fragilise les os et augmente le risque de fracture. C’est pourquoi, plusieurs mesures sont recommandées :
- une alimentation équilibrée en nutriments, minéraux (calcium) et protéine
- une supplémentation en vitamine D, essentielle à l’absorption du calcium
- des exercices de musculation et d’équilibre (marche, course à pied, machines à poids, etc.) pour diminuer le risque de chutes. En cas d’insuffisance respiratoire importante, il est recommandé de pratiquer cette activité sous contrôle médical.
- Voyages aériens : la possibilité de prendre l’avion ou non dépend de la condition respiratoire des patientes. La pressurisation à l’intérieur des avions peut aggraver un pneumothorax pré-existant. C’est pourquoi, le voyage aérien est contre-indiqué en cas de pneumothorax non-traité ou traité dans le mois écoulé. Dans tous les cas, il est conseillé de prendre l’avis d’un médecin spécialiste avant un éventuel voyage en avion.
- Vaccination :il est recommandé de se faire faire vacciner contre la grippe (tous les ans) et contre le pneumocoque (tous les cinq ans) pour réduire les risques de maladies respiratoires.
- Arrêt tabagique (s’il y a lieu) : l’abandon du tabagisme est primordial pour prévenir l’aggravation de la maladie. Il existe plusieurs méthodes pour réussir à cesser de fumer. N’hésitez pas à vous informer auprès d’un professionnel de santé (médecin, pharmacien) à propos de la médication disponible et les thérapies de remplacement de la nicotine.
- Changement dans les habitudes de vie : une activité physique adaptée et une alimentation équilibrée contribuent à atténuer les symptômes de la maladie. Les patientes doivent toujours consulter leur médecin avant d’entreprendre un programme d’exercice ou de modifier leur niveau d’activité physique.
Si vous êtes atteint de LAM
- Connaître les signes de complications pour recourir aux soins en urgence
Toute douleur thoracique ou abdominale soudaine doit faire consulter en urgence et le diagnostic de LAM doit être communiqué à l’équipe soignante
- Le pneumothorax est une urgence médicale. Il se manifeste par une douleur brutale (dite en coup de poignard) au niveau de la poitrine. Cette douleur est majorée par la toux et les mouvements. Si le pneumothorax est important, une difficulté respiratoire (dyspnée) est présente.
- Les angiomyolipomes peuvent se rompre, ce qui provoque de violentes douleurs abdominales et souvent une hémorragie interne dans l’abdomen qui nécessite un traitement d’urgence.
- Effectuer un suivi régulier
Un suivi régulier (plus ou moins rapproché en fonction de l’évolution) est recommandé avec radiographies, épreuves fonctionnelles respiratoires, parfois scanners et gaz du sang. Une échographie abdominale ou une IRM abdominale est généralement recommandée à intervalles réguliers afin de vérifier la présence éventuelle d’angiomyolipomes rénaux ou de surveiller leur évolution.
Une consultation au moins une fois par an dans le centre de référence ou les centres de compétence consacrés aux maladies pulmonaires rares est recommandée. Leurs coordonnées sont disponibles sur les sites Orphanet et RespiFIL.
Relecture par Pr Jacques CADRANEL, Chef de service de pneumologie, coordonnateur du centre de référence constitutif des maladies pulmonaires rares (OrphaLung), hôpital Tenon, AP-HP ; Pr Cécile CHENIVESSE, Cheffe de service de pneumologie et immuno-allergologie, coordinatrice du centre de référence constitutif des maladies pulmonaires rares (OrphaLung), CHU Lille.
