Atelier sur la génétique des Dyskinésies Ciliaires Primitives avec le Dr Marie Legendre – 7 juillet 2021

Organisé par le réseau européen BEAT-PCD*, l’atelier animé par le Dr Marie LEGENDRE, centre de référence des maladies respiratoires rares (RespiRare, Hôpital Armand Trousseau, AP-HP) a réuni de nombreux chercheurs et cliniciens de toute l’Europe autour d’un sujet clé : les approches génétiques actuelles et leurs limites dans le diagnostic de la dyskinésie ciliaire primitive (DCP).
Les dyskinésies ciliaires primitives sont des maladies génétiques rares, de transmission principalement autosomique récessive (il existe de rares cas à transmission liée à l’X ou autosomique dominante).
Dyskinésie ciliaire primitive : hétérogénéité génétique
Si certaines maladies sont dues à des mutations au sein d’un gène unique, de nombreuses maladies génétiques peuvent résulter d’une variation d’un gène parmi un ensemble de gènes plus ou moins vaste. Cette propriété des maladies génétiques est appelée hétérogénéité génétique.
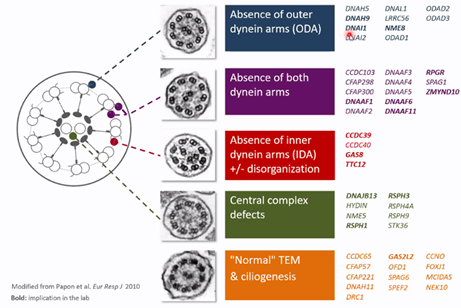
A ce jour, plus de 50 gènes sont impliqués dans la DCP. Cette hétérogénéité génétique explique la diversité des anomalies ciliaires, touchant principalement les bras de dynéine :
- absence isolée des bras de dynéine externes
- absence combinée des bras de dynéine externes et des bras de dynéine internes
- absence des bras de dynéine internes associée à une désorganisation axonémale
- anomalies du complexe central
- absence d’anomalie décelée
- désorganisation axonémale isolée
- diminution des nombres de cils
*BEAT-PCD (Best Experimental Approaches to Treat PCD) est un réseau européen qui réunit des scientifiques, professionnels de santé et associations de patients impliqués dans la prise en charge des dyskinésies ciliaires primitives (DCP).
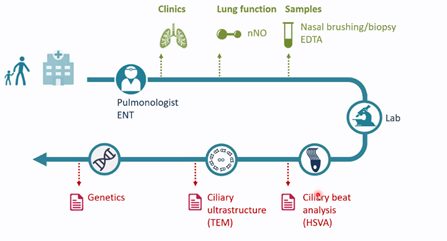
Démarche diagnostique de la dyskinésie ciliaire primitive
Dans le centre de référence des maladies respiratoires rares (RespiRare) à l’hôpital Trousseau (AP-HP), en cas de symptomatologie évocatrice de DCP, des tests fonctionnels tels que la mesure du débit nasal de monoxyde d’azote et/ou l’étude du mouvement ciliaire sont réalisés afin de sélectionner les patients nécessitant une étude de l’ultrastructure ciliaire qui permettra de confirmer le diagnostic de DCP et de guider les analyses moléculaires.
Etant donné que de nombreux gènes et variants responsables de la DCP restent à identifier, cela rend difficile l’utilisation de l’analyse génétique comme test unique pour établir le diagnostic de DCP sans exploration ciliaire préalable. Les résultats des analyses génétiques sont positifs dans environ 22 % de cas de DCP suspectés.
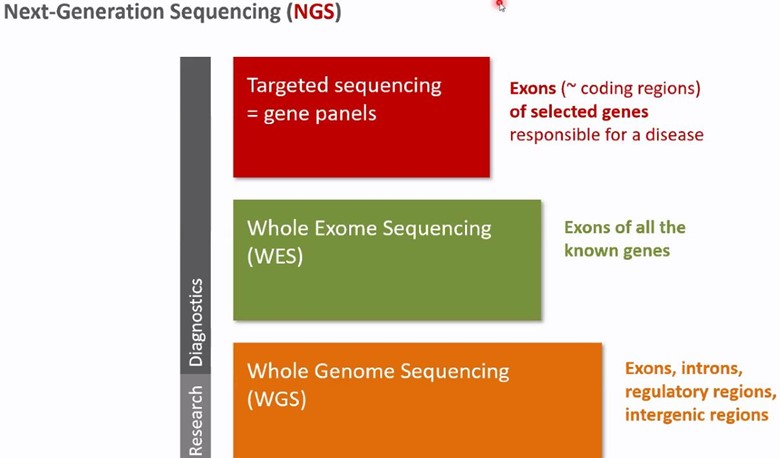
Le séquençage nouvelle génération (Next-Generation Sequencing)
Les tests génétiques reposent principalement sur l’analyse des régions codantes, également appelées exons. La technique la plus fréquente est le séquençage ciblé. Seuls les gènes connus pour être responsables de la maladie recherchée sont alors analysés.
L’analyse du génome entier est également possible, mais cela pose des difficultés pour l’interprétation des données, notamment à cause des nombreuses variations. En effet, il est difficile d’estimer la pathogénicité de certaines d’entre-elles, rares ou non connues.
Les variations génétiques : bénignes ou pathogènes ?
Les variations sont habituellement classées en 5 catégories :
Classe 1 : bénignes
Classe 2 : probablement bénignes
Classe 3 : variations non classifiées
Classe 4 : probablement pathogènes
Classe 5 : pathogènes
Il est fréquent de ne pas pouvoir classifier une variation et cela peut constituer une difficulté puisque la pathogénicité de celle-ci ne sera pas connue. Pour les 3 premières classes, il n’est pas nécessaire de faire du conseil génétique. En revanche, pour les classes 4 et 5 le conseil génétique s’avère plus important car le risque d’être porteur de la maladie est beaucoup plus élevé.

Lorsque l’on séquence l’exome d’un être humain, on observe environ 30000 variations comparativement au génome de référence. Parmi celles-ci, on retrouve 3000 variations rares, ou non-reportées dans les bases de données générales. Environ 300 sont probablement pathogènes et, après avoir enlevé les mutations synonymes, les mutations non-sens et les variations faux-sens, on retrouve entre 10 et 100 mutations pathogènes chez un individu.
Les mutations pathogènes sont fréquentes dans la population générale pour les 51 gènes impliqués dans la DCP. Le risque de développer la maladie est plus important lorsque les deux parents portent la même mutation.
Comment estimer la pathogénicité d’une mutation ?
Plusieurs méthodes sont utilisées pour estimer la pathogénicité de ces variations. Les généticiens se basent, entre autres, sur une analyse de la fréquence du variant dans diverses bases de données de population, la conservation de la base nucléotidique et surtout de l’acide aminé dans l’évolution, la distance physico-chimique entre le résidu aminoacide sauvage et muté, la position dans un domaine fonctionnel connu de la protéine, l’effet de l’épissage (étape de transcription où le gène est amputé de ses introns non codants et où les exons restants sont réunis entre eux).
Il faut être prudent lorsque l’on utilise des outils de prédiction de la pathogénicité. En effet, ces outils tels que SIFT, PolyPhen etc., attribuent un score et une classification en faveur de la probabilité du caractère délétère d’un variant faux-sens ou du caractère bénin du variant en fonction de différents critères cités plus haut. L’effet sur l’épissage est souvent absent et cela peut induire en erreur. Ainsi pour apprécier un éventuel effet sur l’épissage, il est préférable d’approfondir les prédictions au moyen d’outils spécifiques de prédiction des répercussions sur l’épissage.
De nombreuses sources de données sont indispensables à prendre en compte afin d’évaluer une variation génétique. Les outils GnomAD (Genome Aggregation Database) et le UCSC genome browser sont privilégiés et permettent de retrouver la fréquence d’un variant dans les bases de données génomiques.
GnomAD comprend des données issues de 123136 exomes et de 15496 génomes. Il s’agit actuellement de la base de données la plus puissante en termes de nombre d’individus séquencés. En particulier, la présence de 15496 génomes entiers permet d’appréhender la fréquence des variants non-exoniques de manière plus précise. Elle intègre 7 populations différentes.
Le navigateur du génome UCSC genome browser, est une solution permettant d’afficher de manière simultanée plusieurs types de régions complexes en un seul affichage.


