Activités scientifiques conjointes RSRQ-J2R-BeRS : prolifération et hypertension pulmonaire

Les « activités scientifiques conjointes RSRQ-J2R-BeRS » sont une collaboration scientifique conjointe, du Réseau en santé respiratoire du Québec (RSRQ) , des Journées de Recherche Respiratoire (J2R) et de la Belgium Respiratory Society. Le 25 février dernier s’est tenu le premier séminaire virtuel de l’année 2022 autour de la prolifération et l’hypertension pulmonaire. Il a regroupé des présentations avec des orateurs des 3 pays : Belgique, Canada (Québec) et France.
Vous trouverez ci-dessous les communications que nous avons suivies.
France
Traitements actuels de l’hypertension artérielle pulmonaire & innovations thérapeutiques
Maladie pulmonaire rare, l’hypertension artérielle pulmonaire (HTAP) se caractérise par une augmentation de la pression sanguine dans les artères qui relient la partie droite du cœur aux poumons. Cette pathologie résulte d’une prolifération incontrôlée des cellules vasculaires pulmonaires (processus appelé remodelage vasculaire) et une résistance importante à leur apoptose, qui obstruent les petites artères pulmonaires. Comme ce remodelage fait obstacle à l’écoulement du sang dans les vaisseaux des poumons, la pression artérielle pulmonaire augmente. Cette résistance impose un effort au cœur qui à terme peut cesser de fonctionner normalement, et aboutit à une insuffisance cardiaque droite.
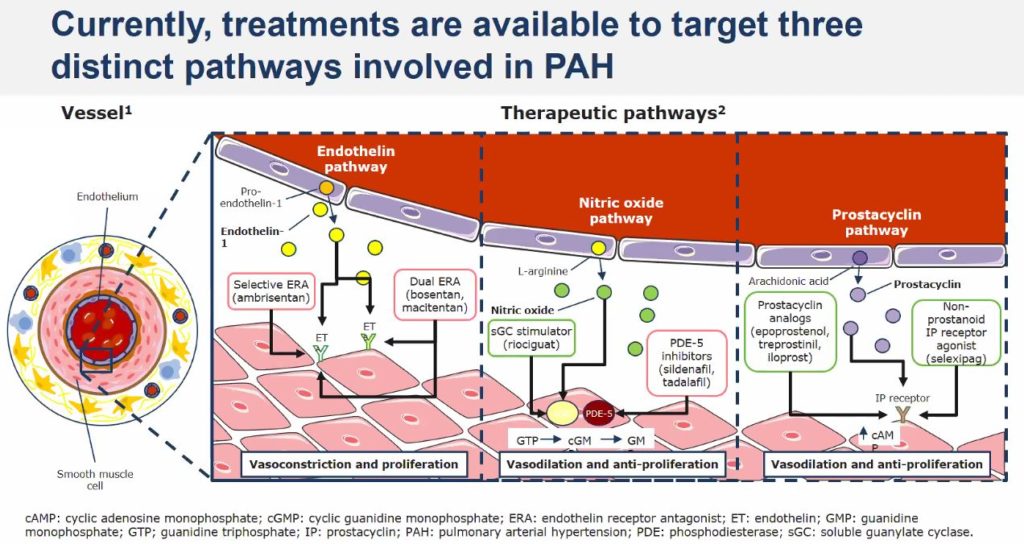
Le Pr Olivier Sitbon (centre de référence coordonnateur de l’hypertension pulmonaire (PulmoTension), CHU Bicêtre,AP-HP) rappelle les médicaments actuellement disponibles dans l’HTAP. Ils ont pour cible l’une des trois voies principales à l’origine de la dysfonction endothéliale artérielle pulmonaire (dont le rôle est avant tout de dilater les artères pulmonaires), impliquant l’endothéline-1, le monoxyde d’azote et la prostacycline :
- Les médicaments qui diminuent l’action de l’endothéline-1 : Bosentan, Ambrisentan et Macitentan
- Les médicaments qui agissent sur la voie du monoxyde d’azote (NO) :
- Inhibiteurs de la phosphodiestérase de type 5 (iPDE‐5) : Sildenafil, Tadalafil
- Stimulateur de la guanylate cyclase soluble (sGC) : Riociguat
- Les médicaments qui agissent sur la voie de la prostacycline (PGI2) :
- PGI2 et analogues : Epoprostenol, Treprostinil,
- Agoniste des récepteurs IP de la PGI2 : Selexipag
« Ces différents traitements en monothérapie ou combinés (ciblant plusieurs voies dysfonctionnelles en même temps) ont montré, dans de larges essais cliniques, une amélioration d’un certain nombre de données cliniques ou fonctionnelles pertinentes, comme les symptômes, les capacités à l’exercice, la qualité de vie, les données hémodynamiques et un ralentissement sur la progression de la maladie. En terme de survie, les traitements combinés associant d’emblée trois médicaments dont une prostacycline ont permis une amélioration significative de la survie », souligne le Pr Olivier Sitbon.
« Les recherches se poursuivent pour améliorer davantage la survie des patients HTAP à long terme », indique l’expert. Ces dernières ciblent des mécanismes différents de la dysfonction endothéliale. « Il est important de souligner que de nombreux traitements ciblant différents mécanismes à l’origine de l’HTAP (stress oxydatif, inflammation, métabolisme, etc.) n’ont pas montré d’efficacité dans des études cliniques ».
Aujourd’hui, de nouvelles thérapeutiques prometteuses, en cours d’évaluation dans des essais thérapeutiques de phase 2 et 3, ciblent les mécanismes potentiellement à l’origine de la prolifération vasculaire pulmonaire tels que :
- la voie du TGF-β : sotatercept, par voie sous-cutanée ;
- les facteurs de croissance comme le PDGF : inhibiteur de tyrosine kinase comme le seralutinib, par voie inhalée) ;
- la voie de la sérotonine : inhibiteur de la tryptophane hydroxylase comme le rodatristat, par voie orale
Focus sur le sotatercept, premier inhibiteur de TGF-β
Le BMPR-II est un récepteur de la superfamille du TGF-β. Lorsqu’elle est activée, la voie du BMPR-II limite la multiplication et l’accumulation des cellules vasculaires dans les parois des artères pulmonaires. Mais lorsque la voie BMPR-II est altérée par des mutations, il s’installe un déséquilibre au profit de la voie du TGF-β et de l’activine qui favorise un remodelage artériel pulmonaire qui est commun à beaucoup de formes d’HTAP.
Dans ce contexte, le sotatercept fonctionne comme un piège à ligands d’un récepteur de l’activine. Cela va permettre de rééquilibrer la balance entre ces deux voies, et ainsi restaurer au moins en partie l’homéostasie vasculaire.
Le sotatercept a montré un effet positif en association aux traitements habituels chez les patients HTAP, à 24 et 48 semaines (étude PULSAR) : amélioration de la distance de marche, réduction de la dyspnée et réduction du marqueur d’insuffisance cardiaque droite (NT-proBNP). Des études de phase III sont en cours » conclut l’expert.
Effet protecteur de l’antagonisme du récepteur des minéralocorticoïdes dans l’hypertension pulmonaire
La pathobiologie de l’HTAP est multifactorielle et concerne potentiellement un dysfonctionnement des cellules musculaires lisses artérielles pulmonaires conduisant à leur prolifération incontrôlée, leur résistance à l’apoptose et in fine à une vasoconstriction. La voie de signalisation minéralocorticoïde joue un rôle important dans la physiopathologie cardiovasculaire. Le récepteur des minéralocorticoïdes (MR), un facteur de transcription, exerce un effet direct sur les réponses prolifératives, pro-inflammatoires et de remodelage. Il peut être activé soit par une voie classique de stimulation dépendante de l’aldostérone, soit par une action directe et rapide de stimulation via le stress oxydatif et/ou via l’activation de l’angiotensine (Ang)-II sur son récepteur AT1 dans le cœur, les poumons et les vaisseaux.
Des études antérieures ont suggéré un rôle de la dérégulation de la voie de signalisation du MR dans le développement et la progression de l’HTAP. Au sein de l’UMR-S999 Inserm/Université Paris-Saclay, les chercheurs ont mis en évidence une augmentation de l’expression (2 à 3 fois) du MR dans les cellules musculaires lisses d’artères pulmonaires (CML-AP) de patients HTAP.
Ainsi, dans ses travaux, le Dr Ly TU (UMR-S999 Inserm/Université Paris-Saclay) a étudié l’impact de l’activation du MR dans les CML-AP et l’efficacité de la finérénone (un antagoniste non-stéroïdien du MR) dans les modèles précliniques d’hypertension pulmonaire.
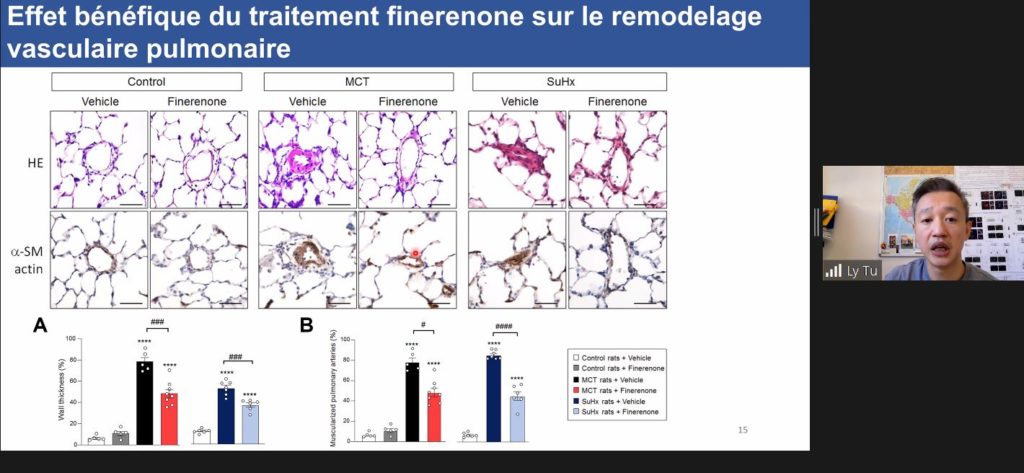
L’analyse des études transcriptomiques* a montré que le MR induit la prolifération des CML-AP, et que les souris sur-exprimant le MR développent une hypertension pulmonaire spontanée, reflétée par une élévation de la pression systolique du ventricule droit, une hypertrophie ventriculaire droite et un remodelage des petites artères pulmonaires.
« En accord avec ces données, nous avons pu observer que l’administration curative de finérénone atténue l’augmentation de la pression artérielle pulmonaire moyenne (PAPm) et améliore le débit cardiaque dans deux modèles complémentaires d’hypertension pulmonaire chez le rat. Enfin, nous avons également constaté que cet effet bénéfique de la finérénone était associé à une baisse de la prolifération des CML-AP et de l’infiltration pulmonaire de cellules inflammatoires» rapporte le Dr Ly TU.
L’ensemble de ces données soutient un rôle du MR dans l’accumulation de CML-AP dans les parois des artères pulmonaires en cas d’HTAP. Ainsi le ciblage pharmacologique du MR pourrait représenter un intérêt potentiel dans le traitement de l’HTAP en conjonction avec les traitements actuels.
*La transcriptomique est l’étude de l’ensemble des ARN (acides ribonucléiques) produits par les cellules et qui révèlent par leur présence la production de certaines protéines, comme les récepteurs par exemple. Ces ARN agissent donc comme des marqueurs intermédiaires.
Québec, Canada
Du rat à la Clinique, inhibiteurs de BRD4 et développement d’une nouvelle approche thérapeutique en hypertension artérielle pulmonaire.
Dans l’hypertension artérielle pulmonaire (HTAP), le remodelage vasculaire est dû principalement à une prolifération anormalement élevée et une résistance importante à l’apoptose des cellules musculaires lisses des artères pulmonaires. Le remodelage vasculaire et la vasoconstriction engendre une obstruction progressive des artères pulmonaires favorisant une augmentation des résistances vasculaires pulmonaires (RVP). Cette augmentation progressive des résistances vasculaires pulmonaires se répercute sur le cœur droit forçant celui-ci à s’hypertrophier pour compenser et maintenir ses fonctions.
Le remodelage vasculaire, étant la conséquence d’une prolifération et d’une survie cellulaire importante, met en exergue une certaine similitude avec le cancer. En effet, certaines voies cellulaires dérégulées dans le cancer, comme ici la prolifération ou la survie cellulaire, se retrouvent également dérégulées dans l’HTAP. La littérature scientifique démontre de plus en plus l’implication de modifications épigénétiques dans le développement de cancer, notamment par la transcription de gènes pro-prolifératifs et anti-apoptotiques. Par ailleurs, les dernières années de recherche ont démontré que des facteurs épigénétiques pouvaient être responsables du développement et de la progression de l’HTAP.
En ce sens, l’équipe de l’Institut Universitaire de Cardiologie et de Pneumologie de Québec/Université Laval (Groupe Hypertension Artérielle Pulmonaire CRIUCPQ) s’est intéressée à la famille des BET (Bromo-and Extra-Terminal) qui comprend quatre membres : BRD2, BRD3, BRD4 et BRDT. Dans des conditions physiologiques, BRD4 est une protéine qui permet d’une part de maintenir la stabilité de la chromatine (support de l’information épigénétique) tout en contrôlant le cycle cellulaire.
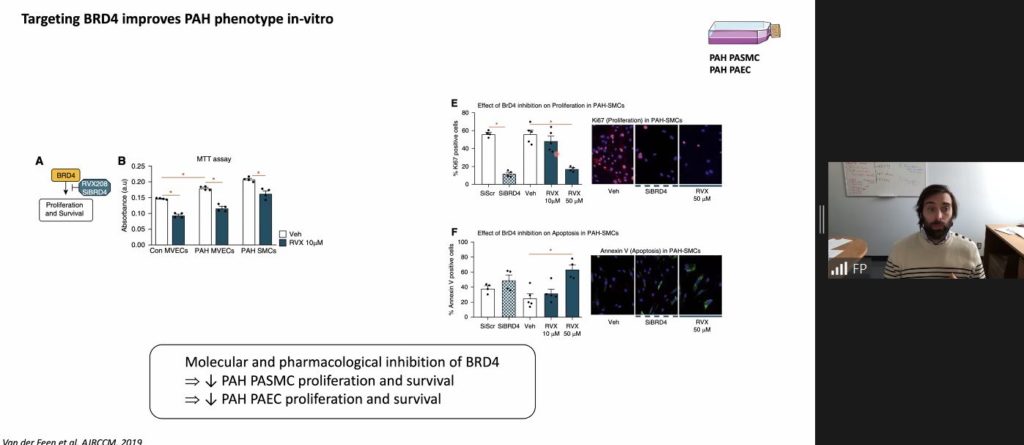
L’étude menée en 2015 par l’équipe du Pr Sébastien Bonnet a démontré que l’expression de BRD4 était augmentée dans les poumons des patients HTAP et plus précisément au niveau des cellules musculaires lisses des artères pulmonaires (CML-AP) de ces patients. De ce fait, BRD4 représente une cible moléculaire favorisant la prolifération et la survie des CML-AP.
Soutenu par ces résultats, une étude préclinique regroupant plusieurs équipes, dont l’équipe de recherche du Pr Bonnet, a mis en évidence l’efficacité du RVX-208, un traitement visant à inhiber la famille des BET dans des modèles expérimentaux de l’HTAP.Ainsi,in vitro, au niveau cellulaire, l’inhibition de BRD4 par le RVX-208 a normalisé l’hyper-prolifération des cellules endothéliales et des cellules musculaires lisses, isolées de patients atteints d’HTAP. De surcroît, Ii vivo, le traitement par voie orale a diminué le remodelage vasculaire et a amélioré les paramètres hémodynamiques pulmonaires chez le rat atteint d’HTAP.
« Étant donnée sa forte implication dans l’HTAP, l’inhibition de BRD4 (RVX208) est une stratégie thérapeutique intéressante qui justifie la mise place d’études cliniques » conclut le Dr François Potus également chercher à l’Institut.
Le Dr Steeve Provencher, du même Institut rappelle qu’à ce jour, les outils thérapeutiques utilisés dans le traitement de l’HTAP ciblent principalement la dysfonction endothéliale en favorisant la vasodilatation. Cependant, cette stratégie thérapeutique n’améliore que de façon médiocre le taux de survie des patients ce qui rend urgent le besoin de développer de nouvelles stratégies ciblant l’obstruction artérielle pulmonaire.
Les résultats positifs et encourageants des études précliniques (citées plus haut) ont donné naissance à une étude clinique visant à tester le potentiel thérapeutique du RVX-208 (nom commercial : apabetalone) chez des patients atteints d’HTAP (NCT03655704).
Cette étude pilote ouverte, à un seul bras, d’une durée de 16 semaines évalue l‘apabetalone (100 mg deux fois par jour) chez les patients recevant déjà un traitement de fond de l’HTAP. Un cathétérisme cardiaque droit et une IRM cardiaque sont pratiqués à l’entrée dans l’étude et après 16 semaines de traitement afin de déterminer les changements dans les paramètres hémodynamiques et la fonction du ventricule droit.

« Les résultats de cette étude démontrent que l’évaluation de l’apabetalone pour le traitement de l’HTAP dans de futures études cliniques (phase 2) est faisable » rapporte le pneumologue. « En effet, tous les participants ont terminé l’étude, sans réduction de dose ni arrêt du traitement. Aucun effet indésirable grave n’a été signalé » rapporte le Provencher.
D’autres études sont nécessaires pour confirmer l’effet bénéfique de l’apabetalone lorsqu’il est ajouté aux traitements actuels de l’HTAP.
Belgique
La perturbation de GCN2 aggrave le remodelage vasculaire au cours de la fibrose pulmonaire
GCN2 est une protéine présente dans toutes les cellules eucaryotes. Il a été montré que GCN2 était impliquée dans la protection contre l’inflammation pulmonaire et la prévention du stress oxydatif et de ses dommages induits par une carence en acides aminés. GCN2 est une kinase dont le rôle est de phosphoryler la protéine eIF2α (eukaryotic initiation factor 2 α). La phosphorylation de eIF2α protège les cellules du stress oxydatif. De façon pertinente pour l’HTP, il a été montré que GCN2 module l’expression de la monoxyde d’azote (NO) synthase (NOS) ; le NO étant un médiateur important du tonus vasculaire pulmonaire.
Dans leurs travaux, les chercheurs ont étudié l’implication de la voie de signalisation GCN2/eIF2α dans le développement de l’hypertension pulmonaire (PH) au cours de la fibrose pulmonaire idiopathique (FPI). Selon la Dr Diana Santos Ribeiro (Université Catholique de Louvain, Bruxelles), « la perturbation de la voie de GCN2 peut contribuer au développement de l’hypertension pulmonaire chez les patients atteints de FPI en altérant la capacité de leur système vasculaire pulmonaire à s’adapter au stress oxydatif ». En analysant le tissu pulmonaire des patients atteints de FPI, les chercheurs ont observé une diminution de l’expression de la protéine GCN2 par rapport aux tissus témoins. De surcroît, l’expression de GCN2 est diminuée dans les cellules endothéliales, indépendamment de l’hypertension pulmonaire. Par ailleurs, les rats traités avec la bléomycine (un médicament anti-cancéreux induisant une fibrose pulmonaire) ont montré une augmentation de la fibrose au niveau du parenchyme et du remodelage vasculaire ainsi qu’une augmentation de la pression artérielle pulmonaire moyenne et une altération de la fonction ventriculaire droite. Comme dans les tissus de patients atteints de FPI, l’expression de la protéine GCN2 a diminué dans les poumons des rats traités par la bléomycine.
Ainsi, ces résultats démontrent que GCN2 est dérégulée à la fois dans la FPI et la FPI associée à l’hypertension pulmonaire.
« La possibilité d’une implication causale de la dérégulation de GCN2 dans le développement de la FPI et/ou de l’hypertension pulmonaire est en cours d’étude » conclut la chercheuse.
L’altération de l’angiogenèse (à l’origine de la résolution des caillots) est un événement clé dans la progression de l’hypertension pulmonaire thromboembolique chronique : validation d’un modèle d’étude de cette maladie
L’hypertension pulmonaire thromboembolique chronique (HTP-TEC) est une complication rare de l’embolie pulmonaire aiguë. La Dr Rozenn Quark (Université Catholique de Louvain, Bruxelles), rappelle les mécanismes qui peuvent entraîner le développement de cette maladie, d’une part, par l’obstruction mécanique des « grosses » artères pulmonaires par des caillots cicatriciels fibreux, et d’autre part, par l’obstruction progressive des « petites » artères pulmonaires entraînant un remodelage vasculaire et un épaississement des parois des artères pulmonaires (semblable à celui retrouvé dans l’HTAP).
« La raison pour laquelle les caillots ne se résolvent pas et entraînent en outre, une obstruction fibro‑thrombotique des artères pulmonaires reste mal comprise. De même, la chronologie des mécanismes qui induisent la persistance de caillots cicatriciels fibreux dans les « grosses » artères pulmonaires n’est pas complétement élucidée. Étant donné qu’une altération de l’angiogenèse est capable d’empêcher la dissolution des caillots cicatriciels fibreux, nous avons cherché à valider son rôle pathogène crucial par l’inhibition in-situ (c’est-à-dire dans les caillots) de l’angiogenèse dans un modèle animal d’HTP‑TEC » indique la chercheuse.
L’équipe a démontré que l’inhibition locale de l’angiogenèse au sein de caillots embolisés à plusieurs reprises chez le lapin induisait une élévation de la pression artérielle pulmonaire moyenne (38 mmHg), une augmentation des résistances vasculaires pulmonaires et une augmentation de l’hypertrophie du cœur droit par rapport à des lapins embolisés avec des caillots contenant un agent antifibrinolytique. Ces résultats permettent de valider un modèle animal reproduisant les principaux aspects physiopathologiques de l’HTP-TEC et offrent des perspectives pour mieux étudier et comprendre les stades précoces du développement de cette maladie rare.



