Essais cliniques
La recherche clinique est une étape essentielle pour mieux comprendre les maladies, améliorer les traitements existants et en développer de nouveaux. Elle repose sur les essais cliniques, menés auprès de volontaires malades ou en bonne santé, dans un cadre strictement encadré pour garantir la sécurité et le respect de chacun. Participer à un essai clinique, c’est contribuer à faire avancer la science et ouvrir la voie à de nouvelles solutions pour mieux soigner les patients.

Comprendre
Un essai clinique, c’est quoi ?
Un essai clinique est une étude menée chez des personnes volontaires. Il permet de tester :
- un nouveau médicament ou une combinaison de médicaments,
- un dispositif médical,
- une nouvelle méthode de diagnostic ou de traitement.
Ces essais concernent à la fois des personnes malades et des personnes en bonne santé. Ils sont très encadrés pour garantir la sécurité et le respect des droits de tous les participants.
Avant de tester un traitement sur l’humain, il est d’abord étudié sur des modèles animaux lors d’une phase appelée pré-clinique. Cela permet de vérifier qu’il est efficace et sans danger.
A quoi servent-ils ?
Les essais cliniques sont une étape indispensable pour :
- commercialiser un nouveau médicament,
- proposer une nouvelle méthode de soins.
Les différents types d’essais cliniques
Deux grandes catégories
- Les essais interventionnels évaluent les effets d’une intervention auprès d’un participant, qu’il s’agisse d’un médicament (efficacité, toxicité), une nouvelle méthode diagnostique ou de dépistage, ou d’un nouveau dispositif médical, etc.
- Les essais non-interventionnels ou observationnels permettent d’améliorer les connaissances d’une maladie et son évolution au cours du temps. Elles sont réalisées dans le cadre du suivi des malades, dans les centres investigateurs.
Qui les organise ?
- Études institutionnelles (ou académiques) : menées par un hôpital ou une université pour améliorer la prise en charge des patients.
- Études industrielles : menées par un laboratoire pharmaceutique pour obtenir l’autorisation de vendre un nouveau médicament.
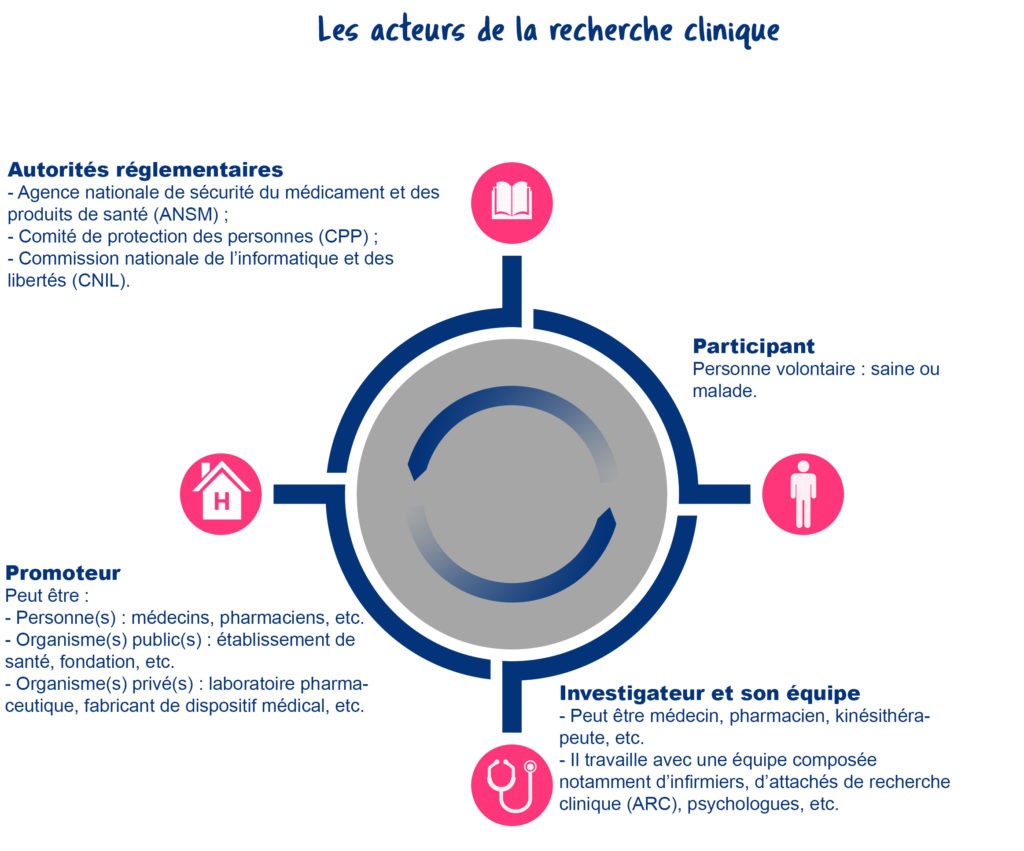
Qui fait quoi ?
- Le promoteur : il organise, finance et suit le bon déroulement de l’étude.
- L’investigateur : c’est le médecin qui encadre l’étude dans un hôpital ou un centre spécialisé.
- Le participant : c’est la personne volontaire. Son engagement est essentiel. Il est libre d’accepter ou de refuser l’étude, et peut se retirer à tout moment.
Les phases d’un essai
Les essais se font en plusieurs étapes (ou phases). À chaque phase, de plus en plus de participants sont inclus.
Dans le cas de maladies rares, comme certaines maladies respiratoires, plusieurs phases peuvent être regroupées (ex : phase I/II ou II/III) pour gagner du temps et inclure plus de patients. Ces essais sont souvent internationaux pour pouvoir réunir suffisamment de volontaires.
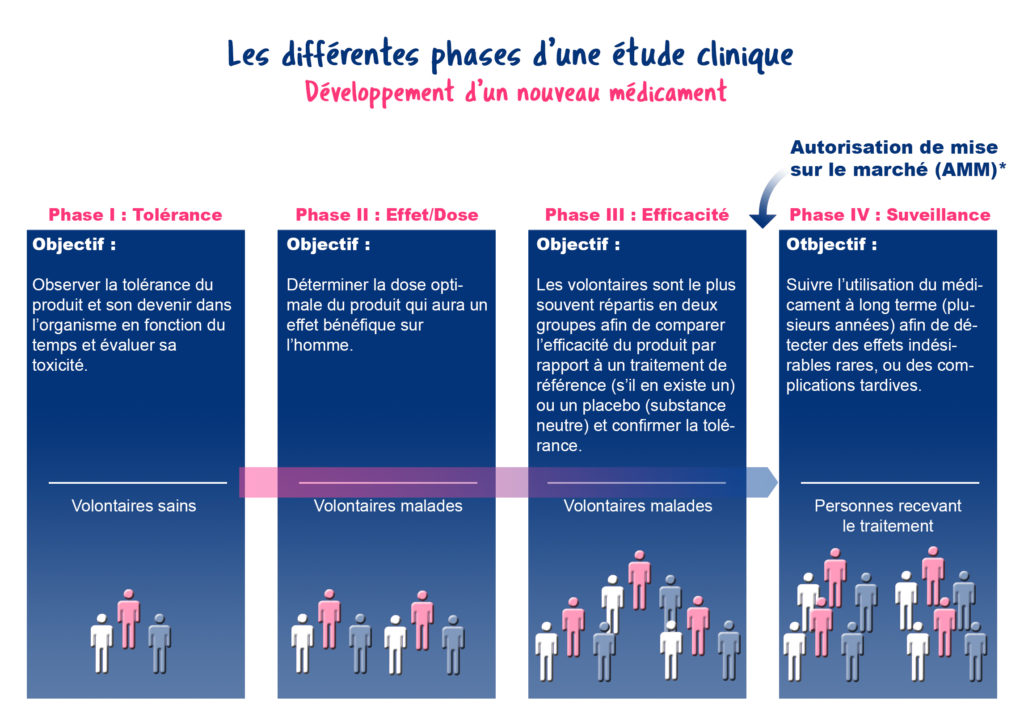
Dans les maladies rares comme les maladies respiratoires rares qui concernent peu de malades, certains essais cliniques sont construits en associant plusieurs phases : on parle d’essais de phase I/II ou II/III. Par exemple, dans un essai de phase I/II, la tolérance sera le critère principal étudié, tandis que les critères secondaires pourront déjà concerner la relation effet/dose, l’efficacité, quant à elle, deviendra le critère principal dans l’essai suivant de phase II/III. Cette façon de faire permet de regrouper suffisamment de personnes pour obtenir des résultats significatifs. Pour les maladies rares, de plus en plus d’essais sont internationaux, afin d’atteindre plus facilement le nombre de participants requis.
Quelques mots à connaître
- Bras : groupe de personnes recevant le même traitement.
- Placebo : produit sans substance active, utilisé pour comparer avec le vrai traitement.
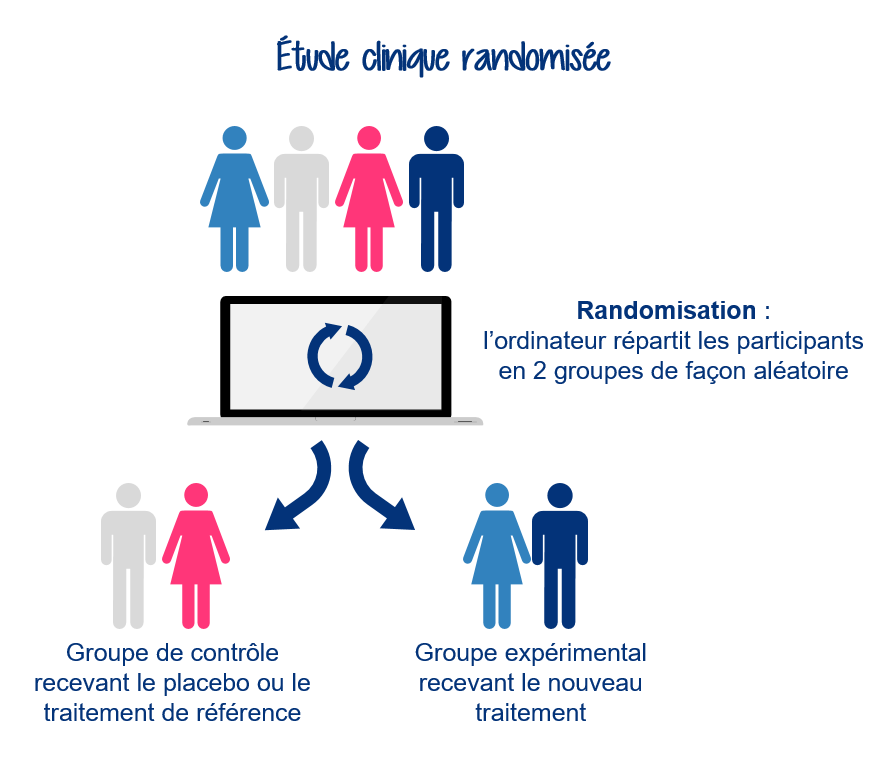
- Randomisation : répartition au hasard des participants dans différents groupes.
- Essai en aveugle : le participant (et parfois le médecin aussi) ne sait pas quel traitement il reçoit pour éviter les biais.
- Essai contrôlé : on compare un groupe qui reçoit le traitement à un autre groupe qui ne le reçoit pas.
Qui peut participer ?
Toute personne majeure, malade ou non, peut participer si elle remplit les critères définis par l’étude. Pour les enfants, l’accord des deux parents (ou du représentant légal) est nécessaire. L’enfant doit aussi être informé selon son âge.
Les critères d’inclusion (âge, maladie, antécédents, etc.) sont vérifiés par le médecin lors d’une visite spéciale dite d’inclusion. Certains examens médicaux sont parfois nécessaires.
Participer à un essai : un choix personnel
Participer à un essai clinique, c’est s’engager dans un protocole rigoureux, pensé pour la sécurité des volontaires. C’est un choix libre, qui ne doit pas être pris à la légère.
Deux documents importants sont remis :
- Une note d’information : elle explique tout sur l’essai (durée, objectifs, risques, bénéfices…).
- Un formulaire de consentement : il est signé si la personne accepte de participer. On peut se retirer de l’essai à tout moment, même après avoir signé.
Pour aller plus loin
Parlez-en à votre médecin.
Contactez les associations de patients ou la filière de santé concernée.
Consultez les sites spécialisés :