Retour sur le CPAP 2024 : le rendez-vous annuel des pneumologues et allergologues pédiatriques

Organisé par l’Association de Pneumo-Pédiatres InterRégionale (ASPPIR), l’Association Immuno-Allergie Infantile (AIAI) et la Société Pédiatrique de Pneumologie (SP2A), le congrès de pneumologie et d’allergologie pédiatrique « CPAP » s’est tenu comme à son habitude à la Maison de la Chimie, Paris 6e du 15 au 16 novembre 2024. Cet événement a rassemblé de nombreux professionnels venus de divers horizons, et a permis d’accroitre la visibilité de la filière des maladies respiratoires rares auprès des pneumopédiatres par la tenue d’un stand dédié.



Plusieurs outils étaient mis à leur disposition, notamment :
- Nouveauté 2024 : les fiches urgences et l’annuaire des PNDS à destination des professionnels pour une meilleure prise en charge diagnostique et thérapeutique des patients, ont été réunis au sein d’une plaquette unique qui permet d’accéder à chacun au moyen d’un simple QRcode ;
- La plaquette des réunions de concertation pluridisciplinaire (RCP) RespiFIL ;
- Le kit transition enfant-adulte Ready Steady Go pour les professionnels de santé concernés par la transition enfant-adulte ;
- Les livrets d’information pour mieux décrire au patient sa maladie et faciliter la compréhension de la prise en charge ;
- Les cartes d’urgence dont l’objectif est l’amélioration de la coordination des soins des patients atteints de maladies respiratoires rares en situation d’urgence ;
- Le livre jeunesse « Lucien est malade », écrit et illustré par la maman de Lucien, un jeune patient atteint de pneumopathie interstitielle diffuse (PID). Ce livre ne mentionne pas la maladie, il peut donc être adapté pour tout jeune enfant devant se rendre à l’hôpital.
De nombreux experts du centre de référence des maladies respiratoires rares – RespiRare étaient au rendez-vous et ont pu présenter leurs travaux. Vous trouverez dans cet article un résumé des communications dédiées spécifiquement aux maladies rares.
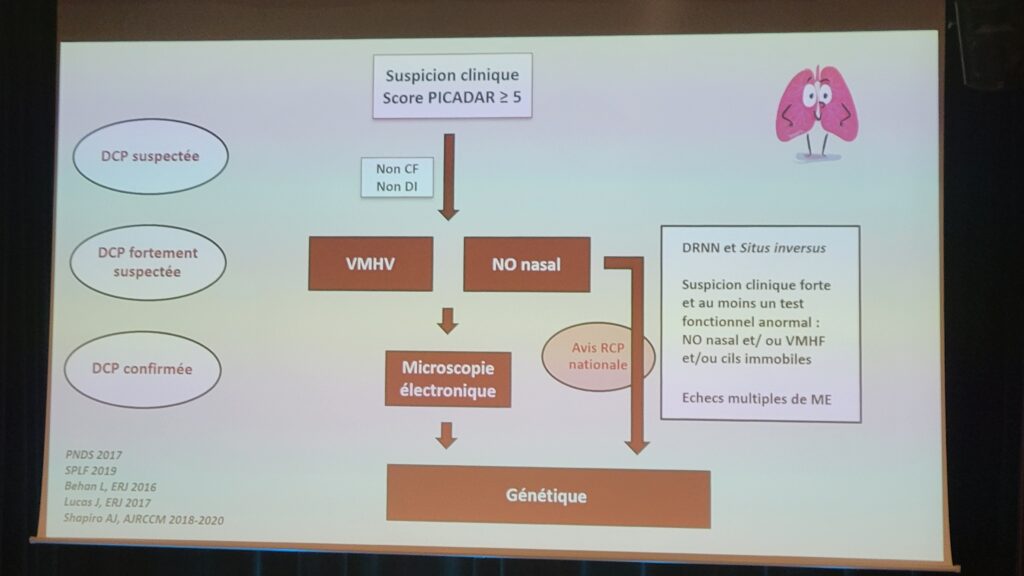
Cette session a permis au Dr Guillaume Thouvenin, pneumopédiatre, à l’hôpital Armand-Trousseau (AP-HP), CRMR constitutif RespiRare, d’informer sur les nouveautés relatives à la dyskinésie ciliaire primitive (DCP) : phénotype, diagnostic, pronostic et prise en charge thérapeutique.
Au sujet des aspects phénotypiques de la pathologie, la nécessité d’une évaluation ORL systématique des patients, de l’intérêt du scanner des rochers ainsi que de l’assistance médicale à la procréation ont pu être discutés. L’intérêt du diagnostic de la DCP par mesure du monoxyde d’azote par voie nasale a également été mentionné ainsi que l’identification de nouveaux marqueurs génétiques liés à la pathologie. À ce sujet, l’algorithme du diagnostic de la DCP a été rappelé avec la nécessité d’user de la rigueur diagnostique adéquate. Enfin, la présentation s’est achevée par l’état des lieux des essais cliniques en cours sur la pathologie, dont : ReCode Therapeutics, Ethris, Parion, Boehringer Ingelheim, INSMED, BEAT-PCD, FOLLOW-PCD, QOL-PCD et CLEAN-PCD, etc..

Les progrès récents de la médecine ont permis de mieux comprendre le rôle des facteurs génétiques dans les maladies pulmonaires interstitielles. Ainsi, des mutations génétiques à forte pénétration ont été identifiées, notamment dans le cas de la pneumopathie interstitielle diffuse (PID) qui touche souvent plusieurs membres d’une même famille.
Cette session a ainsi permis au Dr Alix de Becdelièvre, généticienne à l’hôpital Henri Mondor, Créteil, d’insister sur l’utilité des tests génétiques dans les formes héréditaires de la pathologie facilitant de fait le diagnostic et impactant de manière positive la prise en charge clinique des patients et de leurs proches.
Les tests génétiques dans le cadre de la prise en charge des PID présentent plusieurs objectifs : analyse de l’étiologie de la maladie, la mise en place d’un conseil génétique, une meilleure compréhension de la maladie et la proposition d’une thérapie personnalisée.
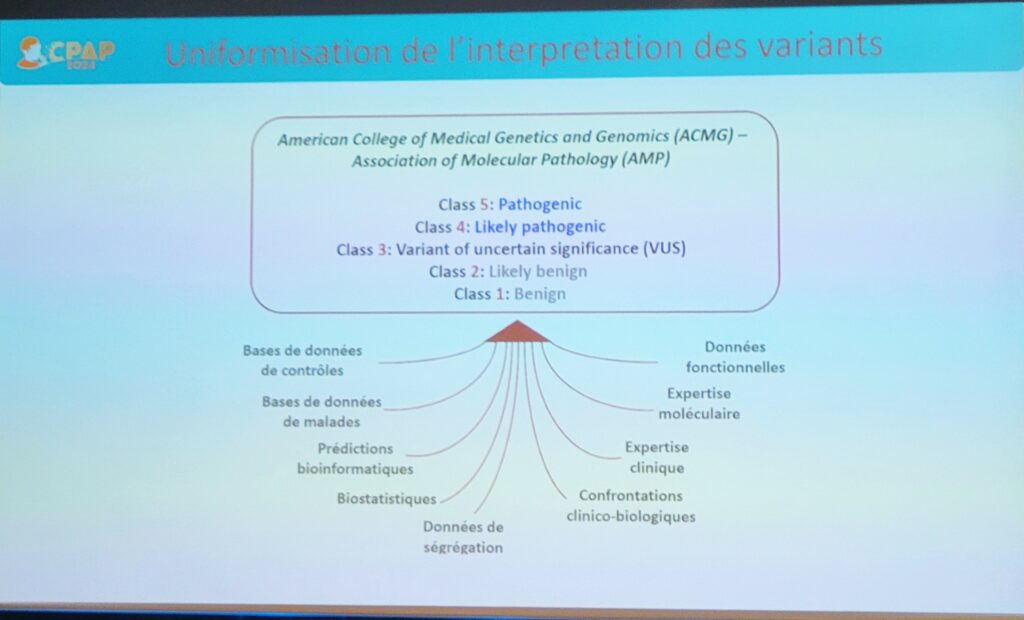
Les nombreux variants génétiques identifiés lors de cette prise en charge sont classés selon 5 classes décrites ci-dessous et selon les dernières recommandations de l’ACMG–AMP (American College of Medical Genetics and Genomics – Association for Molecular Pathology).
- Classe 1 : bénin
- Classe 2 : probablement bénin
- Classe 3 : variant à signification incertaine
- Classe 4 : probablement pathogène
- Classe 5 : pathogène
À ce jour, 45 gènes ont été identifiés comme étant liés à la pathologie dont un nombre conséquent de 300 variants par patient d’où l’importance du phénotypage et du rétro-phénotypage des patients. En cas de résultat positif aux tests génétiques, il est recommandé de mener une étude de ségrégation (dominant/récessif) et d’avoir recours à un conseil génétique.
À noter, qu’un résultat négatif n’exclut pas une maladie d’origine génétique et qu’il illustre plutôt les limites de l’état des connaissances actuelles sur le sujet. L’importance doit ainsi être donnée à la contribution à la recherche sur les variants génétiques par l’alimentation soutenue des bases de données de variants génétiques.
Enfin, la présentation s’est achevée sur la description des dispositions mises en avant par le Plan France Médecine génomique (PFMG) 2025 pour les maladies rares respiratoires. Elles illustrent le soutien des pouvoirs publics vis-à-vis de l’innovation médicale, en l’occurrence du séquençage à très haut débit du génome humain qui fonde la médecine génomique personnalisée.

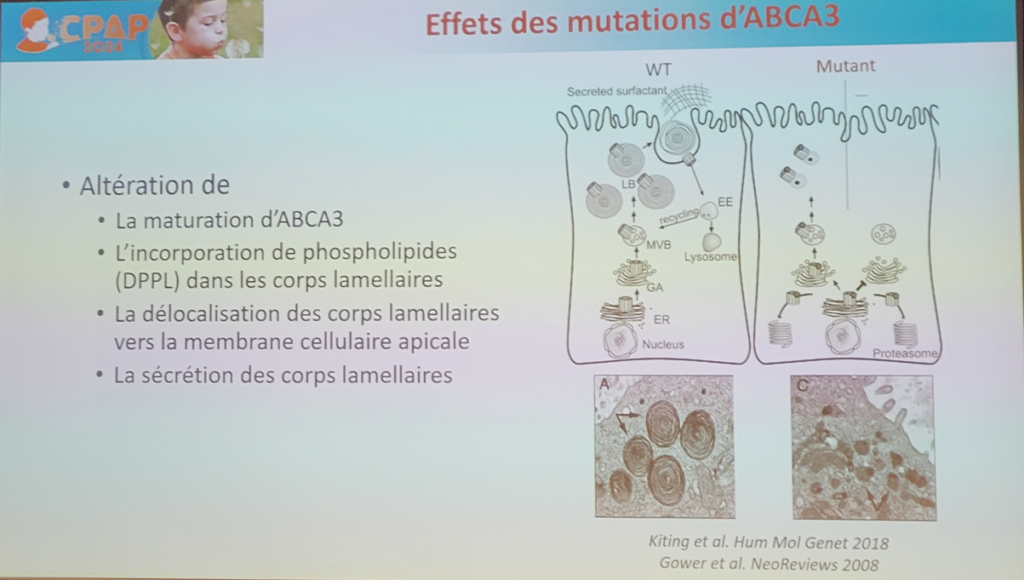
Les pneumopathies interstitielles diffuses (PID) de l’enfant constituent un groupe hétérogène de maladies respiratoires rares chroniques le plus souvent sévères. Parmi elles, la cause génétique est identifiée notamment dans des gènes codant pour les protéines du surfactant (SP)-C (SFTPC), SP-B (SFTPB), leur transporteur ATP-binding cassette, famille 1, membre 3 (ABCA3). La protéine ABCA3 est un transporteur lipidique situé à la périphérie des corps lamellaires dont le rôle est d’incorporer les lipides où s’enchâssent les protéines du surfactant SP-B et SP-C.
La mutation entraine une PID très sévère, rare et à transmission autosomique récessive, avec un début néonatal et une survie à 5 ans de 27 % seulement.
La prise en charge thérapeutique regroupe des bolus de corticoïdes, l’hydroxychloroquine, l’azithromycine, les antifibrosants (nintédanib et pirfénidone) qui ont une autorisation de mise sur le marché chez l’adulte – mais pas chez l’enfant – ainsi que d’autres immunosupresseurs.
Par ailleurs, du fait de l’homologie structurale entre ABCA3 et CFTR, des correcteurs et potentialisateurs de CFTR ont été étudiés et pourraient être une piste thérapeutique intéressante pour les patients porteurs de mutations d’ABCA3 :
- Le lumacaftor est un correcteur du CFTR (maturation et trafic intracellulaire). Une étude a démontré l’effet de l’association de plusieurs molécules bithiazolées et du lumacaftor sur la maturation de l’ABCA3 et sur l’incorporation des lipides mais pas d’effet du lumacaftor seul (Kinting et al, Human Molecular Genetics, 2018).
- L’ivacaftor est un potentialisateur CFTR (augmentation de l’activité membranaire). Une étude a démontré son effet sur l’incorporation des lipides et un effet partiel sur certains variants (Kinting et al, J Cell Mol Med 2019).
- Une étude pilote compassionnelle est portée par le Pr Nadia Nathan, coordonnatrice du CRMR constitutif RespiRare de l’hôpital Armand-Trousseau (AP-HP). Elle vise à déterminer l’efficacité et la sécurité des modulateurs CFTR chez les patients atteints de PID liées au déficit du transporteur ABCA3. Trois patients adultes ont reçu le kaftrio (ivacaftor+ tezacaftor+ élexacaftor). Les résultats ont montré un impact positif pour ces patients par une amélioration de leur état de santé mais des analyses plus poussées seraient nécessaires afin de valider ces premiers résultats.
En conclusion, les modulateurs du CFTR auraient un effet potentiel pour la majorité des patients. Cependant, ils ne sont disponibles qu’en administration compassionnelle, non utilisés chez les nouveaux nés, en l’absence de données sur les effets secondaires à long terme, du manque de soutien des industriels ainsi que la multiplicité des variants bien que des études fonctionnelles soient en cours. Il est rappelé que d’autres perspectives thérapeutiques sont envisageables telles que la transplantation pulmonaire et la thérapie génique.

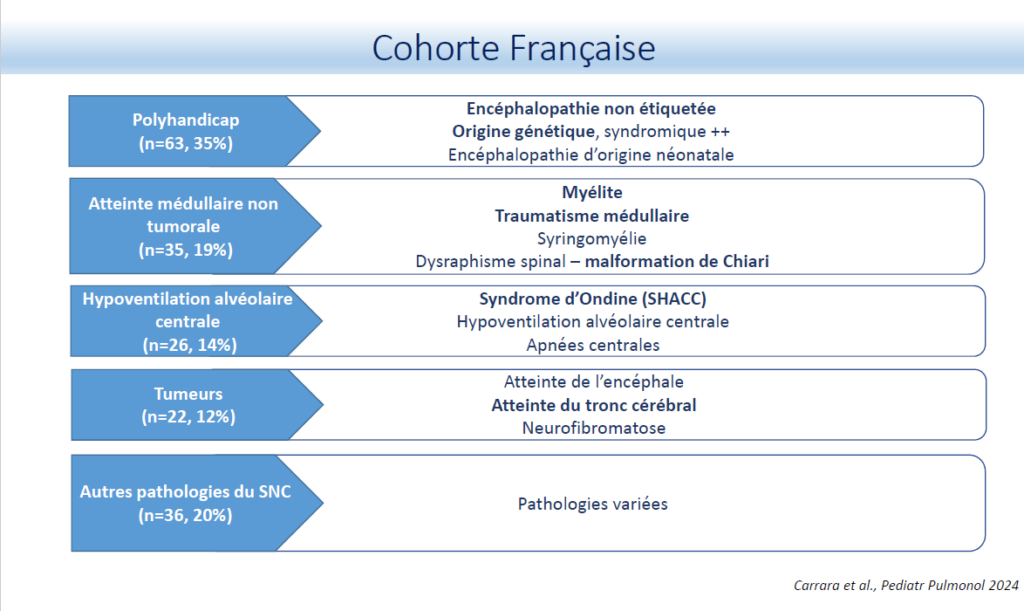
Le Dr Jessica Taytard, pneumopédiatre au service de physiologie – Explorations Fonctionnelles Respiratoires (EFR) – somnologie de l’hôpital Armand-Trousseau (AP-HP), a présenté en premier lieu, les résultats d’une cohorte française de juin 2019 incluant 1447 patients (60 % de garçons), âgés en moyenne de 9,8 ± 5,8 ans dans 28 centres en France. Les troubles sous-jacents les plus fréquents étaient l’obstruction des voies respiratoires supérieures (46 %), les maladies neuromusculaires (28 %), mais aussi une proportion importante des troubles du système nerveux central (13 %). Cette enquête montre le grand nombre d’enfants traités par pression positive continue (PPC)/ ventilation non invasive (VNI) à long terme en France, avec de nombreux enfants présentant des troubles autres que les maladies neuromusculaires. Cette étude a été comparée à une étude rétrospective réalisée en 2003. Une augmentation exponentielle était observée du recours aux supports ventilatoires (PPC/VNI) au long cours chez l’enfant. Entre 2000 et 2019 :
- Le nombre total d’enfants traités par PPC/VNI est passé de 102 à 1447.
- Le nombre d’enfants présentant des pathologies du SNC traités par VNI/PPC est passé de 11 à 182.
- Parmi les 182 patients, 35 % sont atteints de polyhandicaps et sont significativement plus âgés.
- 80 % des patients ont une observance ≥ 4 heures /nuit.
- Observance moins bonne dans le sous-groupes polyhandicap : < 4h/nuit chez 17 % des patients
Limites de l’étude:
- Pas de résultats chiffrés des examens complémentaires pour permettre l’indication de ventilation ;
- Pas d’évaluation des bénéfices : PROMS (qualité de sommeil, qualité de vie), dyspnée, exacerbations et hospitalisations.
VNI et polyhandicaps
Les indications de la ventilation sont toujours complexes du fait de l’absence de consensus de recommandations précises, ainsi que les difficultés d’évaluer les bénéfices attendus sur la qualité de vie. Ainsi, les effets secondaires liés aux interfaces utilisées, aux potentielles inhalations (fausses routes, troubles de la déglutition…) et enfin, la prise en charge globale car il faut prévoir des techniques de désencombrement.
Selon le PNDS de la filière de santé des maladies rares du neurodéveloppement – DéfiScience :
« En cas de repérage de signes cliniques évocateurs, et si le projet de vie du patient s’accorde avec l’éventualité de traitement ventilatoire, des examens complémentaires peuvent être programmés. Ce n’est qu’après avoir investigué toutes les autres solutions possibles que la mise en place du traitement ventilatoire (…) sera éventuellement réalisée. En pratique, cela est rarement réalisable. »
Il existe peu d’études sur la ventilation dans le cas du polyhandicap et dans la plupart des cas, ce sont des études rétrospectives qui s’intéressent à l’observance des patients, la tolérance, les critères d’initiation et l’intérêt de la prise en charge précoce en particulier le désencombrement pour retarder les complications respiratoires.
En conclusion, la mise en place des appareillages de la PPC/VNI a vu une nette augmentation au fil du temps en France. La prise en en charge en cas de polyhandicap est très complexe, d’où la nécessité de poursuivre et multiplier les études sur ce sujet pour uniformiser les recommandations.