En résumé
L’hypertension pulmonaire thromboembolique chronique (HTP-TEC) est une forme d’hypertension pulmonaire survenant après une ou plusieurs embolies pulmonaires. L’embolie pulmonaire est une maladie fréquente. En France, au moins 35 000 cas sont traités chaque année. Chez la plupart des patients, les caillots vont se dissoudre sous traitement anticoagulant, mais dans certains cas rares, la persistance de caillots cicatriciels fibreux dans les artères pulmonaires associée à des anomalies des petits vaisseaux pulmonaires peut conduire à une HTP-TEC.
L’HTP-TEC est caractérisée par une pression artérielle pulmonaire supérieure à 20 millimètres de mercure (mmHg) (la pression moyenne normale est de 14 ± 3 mmHg), des résistances vasculaires pulmonaires (RVP) élevées, une scintigraphie pulmonaire anormale avec la présence d’au moins un défaut de perfusion, une angiographie pulmonaire et/ou un angioscanner thoracique montrant des anomalies caractéristiques de la maladie.
Les raisons pour lesquelles seule une fraction de patients développe une HTP-TEC restent inconnues. Plusieurs mécanismes entraînant une altération de l’élimination des caillots (altération de la fibrinolyse physiologique, activation plaquettaire, angiogenèse déficiente, etc.) pourraient être impliqués. Le résultat final est l’obstruction des artères pulmonaires qui devient permanente, rendant difficile la circulation du sang dans les poumons.
L’HTP-TEC n’est pas une hypertension artérielle pulmonaire (HTAP). Elles constituent 2 groupes distincts dans l’hypertension pulmonaire. Même si les 2 groupes présentent un lien physiopathologique commun ainsi que des symptômes similaires, l’HTP-TEC se différencie de l’HTAP sur de nombreux points (cause, facteurs de risque, diagnostic et prise en charge).
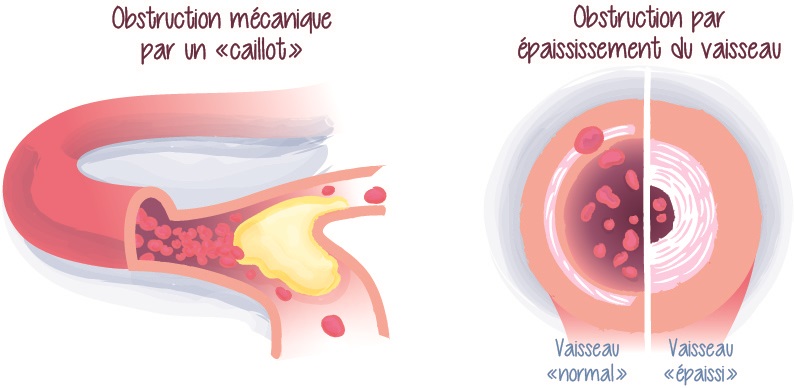
Image 2 : L’obstruction mécanique des artères pulmonaires par des caillots fibreux s’accompagne d’un épaississement de la paroi des artères pulmonaires de plus petites tailles (microscopiques) situées en aval
Personnes concernées
L’HTP-TEC est une maladie rare : sur les 35 000 cas d’embolie pulmonaire survenant chaque année en France, de 0,5 à 1 % se compliquent d’HTP-TEC (soit 300 nouveaux cas annuels et une incidence de l’ordre de 10 par million).
Cette maladie survient autant chez l’homme que chez la femme, plus fréquemment après l’âge de 50 ans. Les formes pédiatriques sont très rares.
Symptômes
Les manifestations cliniques de l’HTP-TEC ne sont pas spécifiques (c’est-à-dire qu’elles sont présentes dans d’autres maladies), puisqu’il s’agit principalement de l’essoufflement (dyspnée) surtout pendant l’effort, qui s’aggrave progressivement. D’autres symptômes peuvent apparaître à mesure que la maladie s’aggrave : fatigue intense, étourdissements ou évanouissement, douleurs à la poitrine, gonflement (œdème) au niveau des chevilles et des jambes.
Diagnostic
Comme les symptômes sont non spécifiques, les médecins réalisent plusieurs examens qui permettront de déterminer s’il s’agit bien d’une HTP-TEC. La détection de l’hypertension pulmonaire repose en particulier sur la réalisation d’une échographie du cœur. Elle permet d’analyser la taille du cœur droit ainsi que le flux sanguin qui y circule par un outil appelé Doppler. Celui-ci permet d’estimer la valeur de la pression dans l’artère pulmonaire. Si l’échographie fait évoquer l’existence d’une hypertension pulmonaire, un cathétérisme cardiaque droit est pratiqué pour confirmer le diagnostic et reconnaître les mécanismes à l’origine de cette maladie.
En cas de suspicion d’une HTP-TEC, des examens d’imagerie du poumon sont réalisés tels que :
- La scintigraphie pulmonaire de ventilation-perfusion pour mesurer l’écoulement de l’air et du sang dans les poumons.
- L’angioscanner pour visualiser et localiser des rétrécissements et/ou des obstructions des artères pulmonaires par des caillots fibreux.
D’autres méthodes d’examens, telles que l’angiographie pulmonaire, peuvent être utilisées pour fournir des informations supplémentaires concernant le diagnostic et l’opérabilité.
Prise en charge
La prise en charge de l’HTP-TEC est multidisciplinaire. En effet, elle nécessite une collaboration entre les experts de différentes disciplines médicales et chirurgicales. Le choix thérapeutique de l’HTP-TEC dépend de la localisation de l’obstruction vasculaire, de la sévérité de l’hypertension pulmonaire évaluée par le cathétérisme cardiaque droit (en particulier la valeur des RVP), et la présence d’autres maladies associées (comorbidités). Ainsi, la prise en charge est adaptée à chaque patient.
Selon la localisation des lésions, certains patients peuvent bénéficier de l’association successive de différentes options thérapeutiques :
- Traitement chirurgical : l’endartériectomie pulmonaire
Elle constitue le traitement de référence quand le patient est opérable, c’est-à-dire lorsque les lésions au niveau des artères pulmonaires sont accessibles par chirurgie. Elle consiste à retirer les caillots fibreux situés dans les grosses artères pulmonaires (obstruction proximale). Ce traitement chirurgical permet de restaurer une fonction cardiorespiratoire proche de la normale pour la majorité des patients opérés. L’endartériectomie pulmonaire est réalisée dans un centre expert (Hôpital Marie Lannelongue au Plessis-Robinson).
- Traitement interventionnel : l’angioplastie (ou dilatation) pulmonaire par ballonnet
Elle permet de dilater les artères pulmonaires de petites tailles qui ne peuvent pas être atteintes par la chirurgie. Elle est donc indiquée chez les patients atteints de formes inopérables (du fait d’une localisation distale des lésions c’est-à-dire sur des artères pulmonaires trop éloignées du cœur droit) ou chez les patients présentant une hypertension pulmonaire résiduelle après chirurgie. Elle permet tout comme la chirurgie de restaurer une fonction cardiorespiratoire proche de la normale chez les patients porteurs d’une HTP-TEC inopérable.
Cette procédure ne peut se faire que dans un centre expert (Hôpital Marie Lannelongue au Plessis-Robinson ou au CHU de Grenoble).
Comment ?
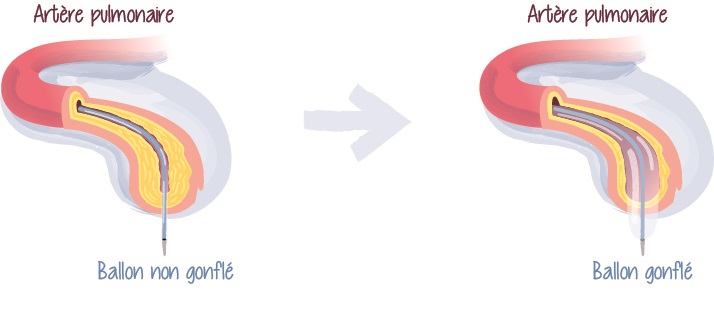
La technique consiste à amener dans l’artère, un petit ballonnet gonflable au niveau de la zone rétrécie. Une fois gonflé, le ballon écrase le caillot sanguin et agrandit le diamètre de l’artère. Il est ensuite dégonflé pour rétablir la voie de la circulation sanguine.
- Traitement médical
Le riociguat est indiqué chez les patients dont les lésions se trouvent dans des artères trop petites pour être accessibles à une endartériectomie pulmonaire. Il est utilisé soit seul soit le plus souvent en association avec l’angioplastie pulmonaire, ainsi que chez les patients présentant une hypertension pulmonaire résiduelle après la chirurgie.
D’autres traitements médicamenteux utilisés dans l’HTAP peuvent également être proposés pour le traitement de l’HTP-TEC avec les mêmes indications que le riociguat (bosentan, ambrisentan, tadalafil, sildénafil, tréprostinil sous-cutané, époprosténol intra-veineux).
Comme dans l’HTAP, certains de ces traitements médicamenteux peuvent être associés si nécessaire.
- Transplantation pulmonaire
Elle est envisagée en cas d’échec des différentes thérapeutiques. Cette situation est exceptionnelle.
Suivi
Après la mise en place d’un traitement pour l’HTP-TEC, il est indispensable d’être suivi tout au long de la vie par une équipe spécialisée. Elle permet de faire le point, à intervalles réguliers, sur :
- L’efficacité et l’observance des traitements
- L’apparition ou non de nouveaux symptômes
- La gestion des symptômes et du mode de vie
Les outils utilisés pour le suivi comportent :
- L’évaluation des symptômes (importance de l’essoufflement à l’effort)
- Le test de marche de 6 minutes. Il mesure la distance que la personne peut parcourir en marchant pendant 6 minutes.
- Le dosage sanguin de certaines substances sécrétées par le cœur (BNP ou NT-proBNP)
- Le cathétérisme cardiaque droit
- L’échographie cardiaque
Liens utiles
Informations pour tout public
Informations pour les professionnels
Relecture par le Dr Xavier Jaïs, pneumologue, centre de référence coordonnateur de l’hypertension pulmonaire (PulmoTension), hôpital Bicêtre – AP-HP, Le Kremlin-Bicêtre.