Retour sur l’atelier collaboratif DAAT

À l’initiative de l’association nationale française ADAAT Alpha1-France, le 21 février dernier s’est tenu l’atelier collaboratif sur le déficit en alpha1-antitrypsine (DAAT) réunissant cliniciens, chercheurs, laboratoires pharmaceutiques et RespiFil autour des membres de l’association.
Le déficit en alpha1-antitrypsine est une maladie génétique causée par des variations du gène SERPINA1 qui produit la protéine A1AT dans le foie. Cette protéine doit normalement migrer par le sang et agir dans les poumons. Elle doit alors détruire les protéines enzymatiques (protéases, élastases) des globules blancs chargées de tuer les microbes lorsqu’elles ont fini leur travail. En son absence, les protéases et élastases vont s’accumuler ce qui peut abimer les alvéoles des poumons, créer de l’emphysème.
Les objectifs de cet atelier étaient d’augmenter les interactions entre les experts, d’améliorer la prise en charge, le parcours de soin sur l’ensemble du territoire, renforcer les programmes de recherche et la communication à destination des patients et, développer la réflexion autour de la transition enfant-adulte.
Vous trouverez ici, les résumés des sessions :
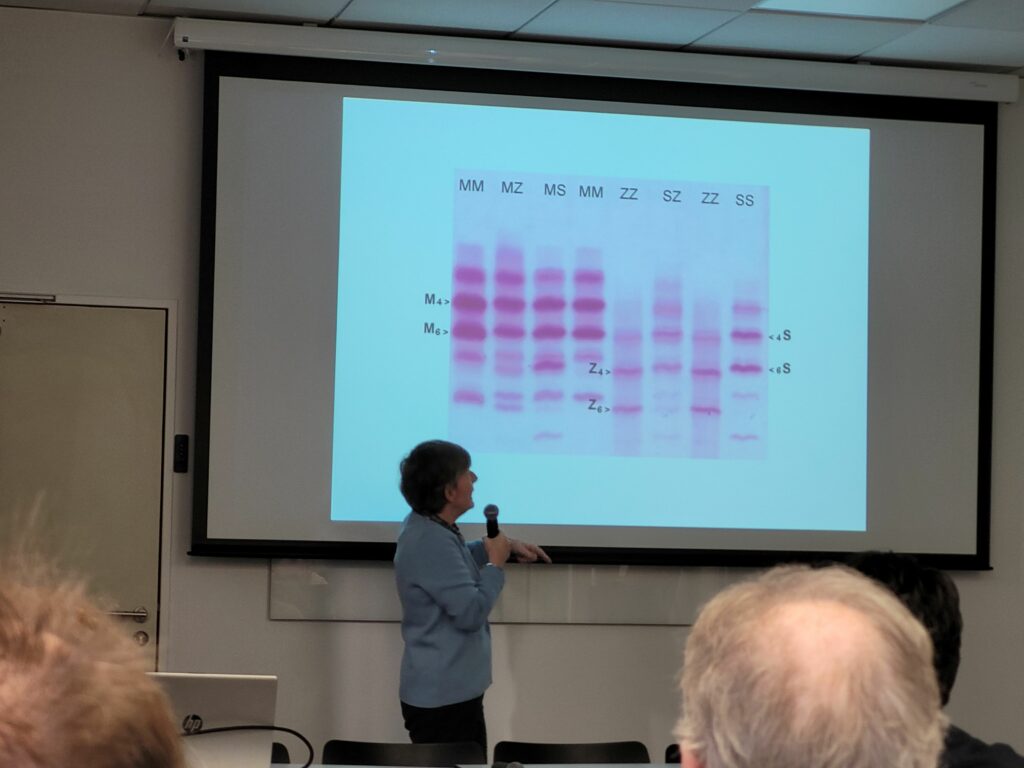
Le Dr LACAILLE (hôpital Necker-enfants malades, filière FILFOIE) a tout d’abord présenté l’isoélectrophorèse utilisée pour préciser les variants génétiques du gène SERPINA1, notamment les variant S et Z associés au déficit en alpha1-antitrypsine.Trois centre de référence en France maitrisent cette technique : Lille, Lyon et Paris. Les gènes normaux sont nommés M. Une personne qui n’est pas atteinte d’un DAAT possède deux gènes M (MM). Les personnes atteintes d’un déficit en A1AT ont le plus souvent deux gènes Z (ZZ).
Dans les cellules du foie, la protéine A1AT se replie mal, elle se met en boule, en pelote en faisant des nœuds et ne peut pas être transportée dans la cellule pour être relarguée dans le sang. Il faut donc pouvoir l’éliminer dans la cellule avec différents mécanismes. Il existe une variabilité entre individu qui dépend de l’efficacité de chacun à éliminer l’A1AT. À défaut, le foie peut également finir par être malade mais l’évolution est généralement lente. Le DAAT reste une indication très rare de greffe hépatique chez l’enfant (cirrhose < 3 % avant 20 ans). Par contre, les enfants peuvent plus fréquemment développer une hypertension portale car le sang doit circuler en faisant le tour du foie.
Le Dr MILORD, hépatologue (hôpital Beaujon, filière FILFOIE), rappelle que le DAAT est une maladie rare, sous diagnostiquée (< 20 % des personnes atteintes sont diagnostiquées), dont la prévalence varie selon les sources d’1/2400 à 6000 personnes. Elle toucherait plus les populations d’Europe du Nord.

Au niveau du foie, de la physiopathologie, l’A1AT s’accumule ce qui crée une toxicité en interne avec un risque plus accru pour les patients ZZ. Les hétérozygotes, SZ et, MZ, ont également un risque augmenté d’avoir une atteinte hépatique.
Les cellules du foie entrent en activité pro-apototique pour éliminer la protéine anormale et se régénèrent ce qui crée une inflammation, de la fibrose, des cicatrices qui peuvent évoluer en cirrhose et cancer du foie.
Souvent les patients très graves ont des cofacteurs – épigénétique, environnement, habitudes de vie (alcool, surpoids, obésité, etc.) – qui donnent une atteinte du foie plus sévère. Parfois de petites anomalies de l’analyse de sang, comme une légère augmentation des transaminases, retardent le diagnostic car cela peut être mis sur le compte d’une infection, de la consommation d’alcool, de l’obésité. Il faut donc suivre les chiffres sur plusieurs mois. Après 50 ans, les suivis sont rapprochés.
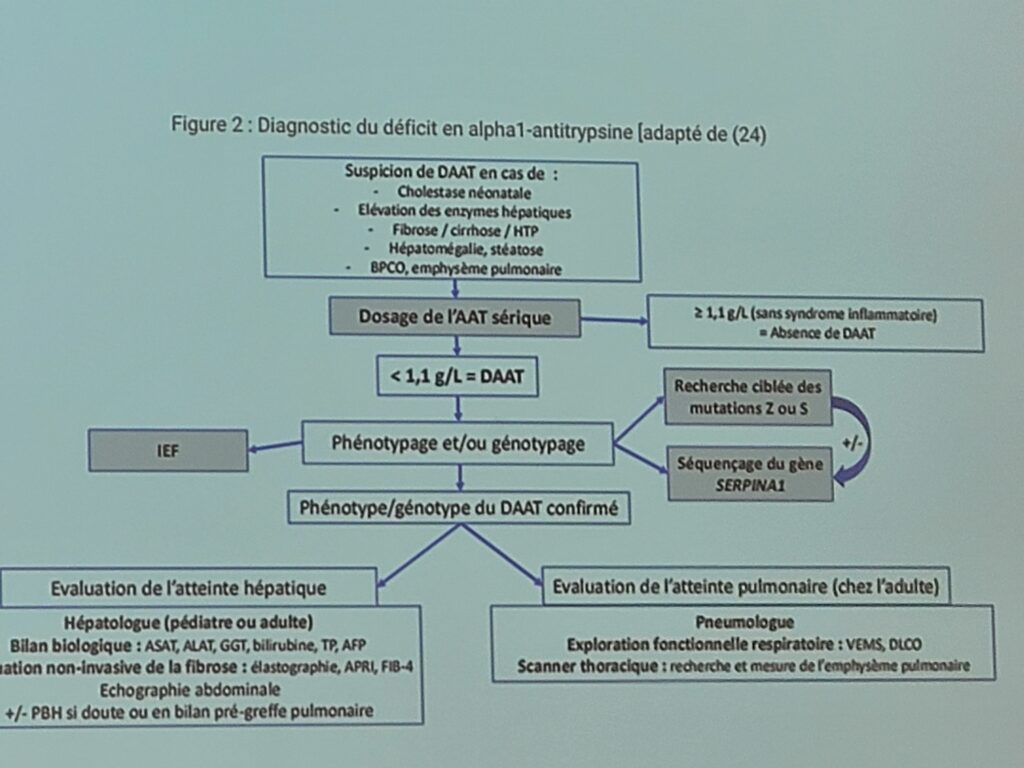
Comment faire le diagnostic ? Si les patients ont une atteinte pulmonaire souvent le diagnostic est déjà fait lors de l’arrivée en hépatologie. Sinon, il faut interroger le patient sur son enfance, son parcours, une imagerie anormale/atypique et faire simplement un dosage sérique dans le sang (protéines inflammatoires). Le phénotype/génotype est assez rapidement pratiqué comme le fibroscan (non invasif). Cela donne des informations pour savoir si la biopsie (invasive) pourrait être bénéfique.

L’atteinte pulmonaire est liée à un déséquilibre entre la protection pulmonaire assurée par l’alpha1-antitrypsine et la charge à l’agression pulmonaire par les protéases induite par les infections, agressions dont le tabagisme. Plus le taux d’A1AT est bas plus la perte de protection pulmonaire est importante.
En France, 10 000 patients seraient homozygotes (ZZ), il existe donc un réel sous diagnostic. Les patients ZZ sont les patients à risque le plus important, comme les Q0 (absence totale de protéine). Le développement de la pathologie pulmonaire est conditionné certes par le déficit mais aussi par la présence d’agression dont le tabac qui joue un grand rôle.
Le Dr LE ROUZIC présente ensuite les recommandations françaises du Pr Jean-François MORNEX publiées en 2022 dont l’impact de l’exposition au tabac sur la survie. Il n’existe donc pas de fatalité et il recommandé de ne pas fumer.


Quand les patients ont des variants rares, peu connus, ni S, ni Z, ni M, il est proposé de discuter du risque pulmonaire et hépatique lors de la réunion de concertation pluridisciplinaire (RCP) car ces patients ne figurent pas dans les recommandations.



Dans l’atteinte pulmonaire, il est possible de recourir à un traitement substitutif et donc d’apporter l’alpha1-antitrypsine manquante pour rétablir la protection pulmonaire grâce à un apport intraveineux par perfusion hebdomadaire de la protéine extraite de dons du sang. L’intérêt du traitement substitutif est de ralentir le déclin pulmonaire, l’emphysème, l’effet est donc modeste car il ne répare pas le poumon et ne réduit pas l’essoufflement. Par ailleurs, il n’existe pas de critère défini pour arrêter ce traitement.
Chargée de recherche dans l’équipe BioGO au BRIC, Bordeaux, le Dr BOUCHECAREILH a dressé l’inventaire des avancées thérapeutiques basé sur la compréhension de la physiopathologie pour identifier de nouveaux outils thérapeutiques en dehors du traitement substitutif issu de don de plasma.

Citons :
- L’édition du génome par la stratégie phare du CrispR/Cas9 : « ciseaux moléculaires » qui vont couper l’ADN précisément par coupure sur le gène SERPINA1 puis le réparer en insérant le correctif. Deux laboratoires sont en phase de développement, Beam Therapeutics avec le Bean-302 et Prime Medecine.
- L’édition réversible de l’ARN : approche basée sur l’enzyme ADAR impliquent plusieurs sociétés biotechnologiques.
- ARN interférent (siRNA) qui vise à dégrader l’ARN message du mutant Z et ainsi réduire la synthèse de la protéine anormale. Le Farzisiran est développé par Takeda.
- Le repliement de la protéine avec des folding correcteurs par BioMarin avec le BMN349 et VERTEX avec le VX-864 qui présentaient tous les deux de bons résultats précliniques. Ils ont cependant dû être stoppés en essai clinique.
- Le remplacement de la protéine avec de l’AAT recombinante comme le propose notamment Sanofi avec l’Efdoralprin alfa (INBRX-101) actuellement en phase 2 avec la désignation de médicament orphelin à l’EMA. Mereo BioPharma développe un inhibiteur des élastases neutrophiles. Il s’agit de l’alvelestat qui passe également en phase 2.
En conclusion, l’innovation, la conception des études et la mise en œuvre de thérapies émergentes pour le déficit en AAT sont autant de raisons d’être optimistes.
- L’AAT recombinante pourrait réduire les coûts financiers et la dépendance au don de plasma.
- La thérapie par ARN interférent (RNAi) offre une approche préventive contre les atteintes hépatiques. C’est la thérapie la plus avancées à l’heure actuelle.
- L’édition de l’ARN et la thérapie génique vont encore plus loin avec l’obectif de guérir la maladie en permettant la production d’AAT fonctionnelle directement dans le foie natif.
L’association ADAAT Alpha1-France fêtera ses 20 ans en 2027 ! Mme LEFRANÇOIS, sa présidente, présente les différentes activités :
- Journée des familles : activité physique adaptée (APA), yoga, atelier culinaire, etc.
- APA en visioconférence : 3 groupes par semaine. Maximum 8 par personne.
- Sophrologie à distance, en illimité accès sur la respiration puis la relaxation
- Livret pour les adolescents, jeu pédagogique à développer
- Café alpha en région ouvert aux non membres également, dates sur Facebook

- Sensibilisation des praticiens hospitaliers
- Travail sur des capsules vidéos pour sensibiliser et informer
- Webinaires thématiques tous les 2 mois, lien sur Facebook
- Deux applications Alpha & Moi (Application Peach, soutenus par les laboratoires GRIFOLS et LFB) et My alpha Compass (CSL Behring) plus adapté pour le suivi des perfusions
- Défense des droits des patients avec le Collectif droits à respirer (CDAR) et Alliance Plasma
- Soutien aux patients dont le fond social
- Soutien à la recherche : 20k€/an pour le Dr BOUCHECAREILH et l’hébergement de la cohorte DEFIALPHA.
M. Mathias SICAUD, pharmacien, cofondateur de la société Peach spécialisée dans le développement de compagnons digitaux personnalisés et adaptés à chaque pathologie en co-construction avec différents partenaires : association de patients, centres experts, filières, etc. et donc ceux qui vivent et suivent la maladie. Ce compagnon, gratuit, doit être l’allié du patient.

Le compagnon « alpha et moi » a pu être développés grâce au soutien institutionnel des laboratoires GRIFOLS et LFB. En moins de 10 min, il est configuré directement par le patient qui peut y ajouter l’ensemble de ses traitements. Le compagnon permet également le suivi des symptômes (échelle d’évaluation), des exacerbations, de préparer la consultation (rétroplanning, examens à faire), d’inclure des questionnaires et de permettre de poser des questions aux médecins. À l’issue du remplissage, un .pdf est généré et peut être partagé dans l’espace santé mais aussi l’imprimer.

Cette journée a permis à l’ensemble des parties prenantes de s’exprimer, de partager ses connaissances et ainsi d’initier des pistes de travail commun. Le groupe de travail collaboratif se poursuivra donc sur les prochaines années. Merci encore de nous y avoir convié !


