Colloque annuel 2024 de la Fondation Maladies Rares

La Fondation Maladies Rares est une fondation de coopération scientifique à but non lucratif ayant comme objectif de favoriser la conduite de projets de recherche et d’excellence scientifique ainsi que le partage et la diffusion des connaissances dans le domaine des maladies rares.
Le 18 juin 2024 s’est tenu son colloque scientifique annuel, axé sur la recherche, au campus des Cordeliers à Paris. Cette journée, placée « Au cœur des progrès et des défis de recherche sur les maladies rares », a été introduite par le Pr Jean‑Louis Mandel, président de la Fondation Maladies Rares. Elle a mis en lumière les progrès de la recherche avec différentes sessions et focus alors que les temps informels permettaient d’échanger avec les partenaires institutionnels et les professionnels dont les posters étaient présentés sous le cloître.
La matinée d’échanges à laquelle nous avons pu assister a regroupé 250 participants en mode hybride. Elle a été riche en enseignement et ouvre un champ d’espoir pour l’amélioration de la prise en charge des patients notamment grâce aux sciences humaines et sociales.
DIAGNOSTIC ET AVANCÉES DANS LA COMPRÉHENSION DES MALADIES RARES
Ce qu’il faut retenir :


La génétique de la fertilité connaît de grandes avancées ces dernières années avec notamment le développement de techniques de séquençage à haut débit qui révolutionnent la pratique du diagnostic de l’infertilité et modifient la prise en charge des patients. Ainsi :
- 30 à 40 % des infertilités pourraient être de causes génétiques ;
- 2 formes : syndromiques comme dans la myotonie de Steinert, la mucoviscidose ou les ciliopathies (dont la dyskinésie ciliaire primitive) avec défaut du flagelle du spermatozoïde, et non syndromiques avec des statuts de fertilité non étudiés ;
- Les gènes recensés qui concernent les 4 phénotypes d’infertilité féminine nécessitent une prise en charge particulière de ces patientes (préservation ou don d’ovocytes, FIV selon les situations) :
- Insuffisance ovarienne prématurée (IOP)
- Défaut de maturité ovocytaire
- Échec de fécondation
- Défauts / létalités embryonnaires préimplantatoires
- Pour les gènes impliqués dans l’infertilité masculine, là encore, il faut orienter, selon les situations, vers la préservation, le don de sperme ou la FIV ICSI.
« En quelques années de recherche, nous sommes passés de 16 gènes au dernier panel à 179 gènes, et maintenant, avec l’exome, le panel in silico contiendra 300 gènes. »
Le nouveau paradigme concerne le conseil génétique des patients mais aussi des proches qui seraient porteurs d’anomalie génétique. Bien que cela paraisse évident pour des généticiens, ce message doit passer dans les centres de fécondation in vitro auprès des médecins de la reproduction.


Ce qu’il faut retenir :
- Le développement sexuel est un processus extrêmement complexe dont l’ensemble des acteurs n’est pas connu.
- Les personnes présentant des variations du développement génital (VDG) pour la moitié d’entre-elles n’ont pas de diagnostic génétique ce qui peut avoir un impact important sur leur prise en charge
- Le but du projet est de confirmer l’implication de gènes déjà connus ou de confirmer des gènes émergents décrits avec peu de cas voire d’identifier de nouveaux gènes.
- Cette étude prospective menée sur le CHU de Rennes a concerné 10 fœtus (5 fœtus XX, 5 fœtus XY) présentant une VDG syndromique. Elle a permis d’identifier des variants pathogènes déjà connus MYRF, PORCN et AMH ainsi que 3 autres gènes connus en pathologies humaines mais dans lesquels les VDG n’étaient pas décrites : MMP21 (se rapproche d’une ciliopathie), PP2R1A. Le gène SLC25A26, lui, est récemment décrit surtout chez des patients adultes mais aussi chez des enfants atteints d’HTAP.
- L’exome en trio permet un apport diagnostique de 60 % ce qui est comparable aux données retrouvées dans la littérature tout en confirmant l’implication de gènes connus ou d’étendre leur phénotype pour un coût et délai de rendu intéressants.
- La principale problématique concerne la mise en place de test fonctionnel chez des fœtus décédés, la corrélation phénotype/génotype avec la littérature qui décrit les cas les plus sévères, les atteintes d’organes les plus sévères, sans que les VDG soient toujours rapportées. Enfin, l’inconvénient majeur réside dans l’analyse du génome non codant (UTR, régions régulatrices).
Ce qu’il faut retenir :
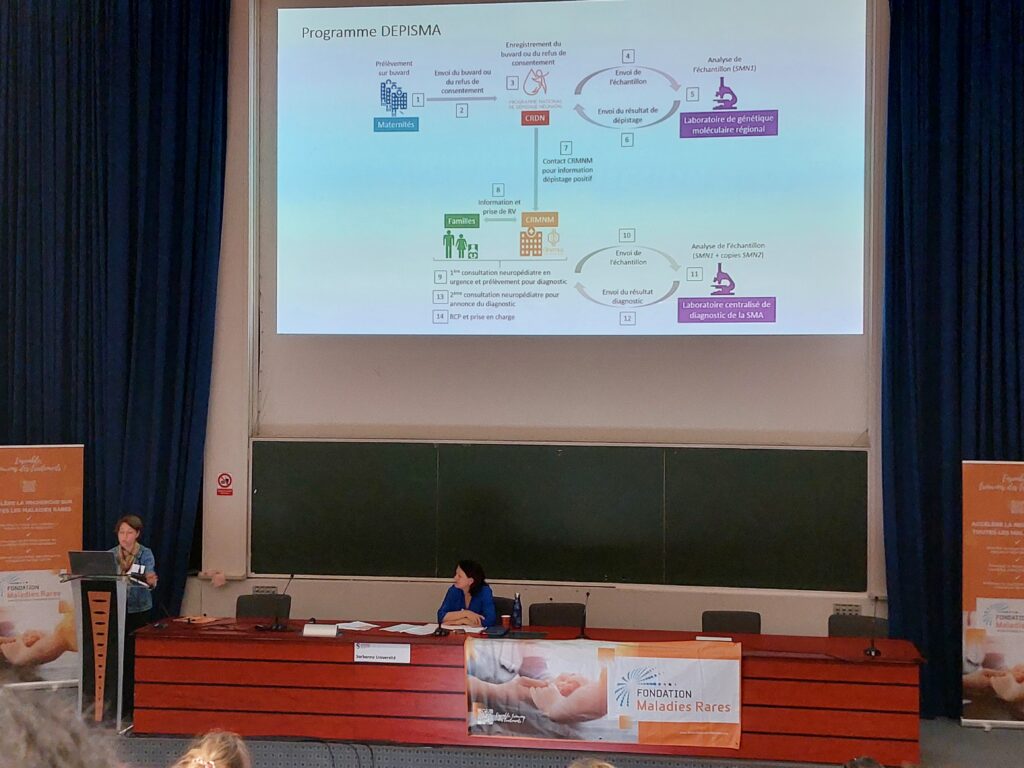

- L’amyotrophie spinale est une maladie du motoneurone à transmission autosomique récessive qui touche 1/7000 naissances soit environ 100 nouveaux cas par an en France. Elle conduit à une paralysie musculaire progressive. Dans 95 % des cas, les patients sont porteurs d’une mutation homozygote du gène SMN1. La forme 0, la plus sévère, est la plus précoce alors que la forme la moins sévère, la plus tardive est la forme 4.
- Il existe une corrélation inverse entre la sévérité du phénotype et le nombre de copies de SMN2 qui codent un peu pour de la protéine fonctionnelle.
- Depuis 2017, des traitements ont révolutionné la prise en charge des patients d’autant plus qu’ils sont administrés précocement. Le choix de l’orientation thérapeutique se fait dans le cadre d’une réunion de concertation pluridisciplinaire nationale.
- Actuellement pour le type I, le plus fréquent, 40 % des enfants sont diagnostiqués trop tard ce qui justifie la mise en place d’un dépistage néonatal qui fait consensus au niveau international depuis 2020 (USA, Canada, Europe dont l’Ukraine). D’où ce projet préfigurateur en France où le dépistage est basé sur des techniques de biologie moléculaire en première intention grâce à la Loi de bioéthique 2022.
- L’objectif du projet DEPISMA est de démontrer la faisabilité de ce dépistage en France pour des enfants nés dans les régions Grand Est et Nouvelle Aquitaine, soit 82 maternités concernées, pendant 2 ans, avec des buvards comme les tests actuels.
- Le projet a débuté fin 2022 à Strasbourg et en 2023 en Nouvelle Aquitaine pour près de 95000 nouveau-nés testés avec un taux de refus proche de 1,5 %. Le délai de résultat est de 7 jours. Sept résultats se sont avérés positifs, soit une incidence de 1/11881, dont 5 nouveau-nés sont traités pas thérapie génique.
- Cette étude prouve la faisabilité de ce dépistage néonatal comme l’acceptabilité par les parents. Les algorithmes biologiques et cliniques sont prêts. Ainsi DEPISMA ouvre la voie à un programme national de dépistage néonatal de l’amyotrophie spinale.
Ce qu’il faut retenir :

- Les mutations du gène WNT10A impliquent des défauts de synthèses de protéines qui posent des problèmes dans l’ostéogénèse avec des hypotrophies osseuses importantes chez les jeunes patients.
- Avec le Pr François Clauss, l’objectif est de développer des traitements thérapeutiques pour ces patients avec des défauts de la mâchoire, des hypotrophies et des oligodonties notamment avec la mise en place de modèles expérimentaux avec des membranes PCL (fibreuses avec une bonne intégration cellulaire, facilement implantable) qui permettent la régénération osseuse. L’idée est de mobiliser les outils de formulation pour développer un dispositif stimulant pour les patients atteints d’oligodontie wnt10a. Ainsi, la reconstruction mandibulaire, bioactive (ostéogène et angiogène pour la calcification et la vascularisation), pourrait servir de support à l’implantation dentaire avec une procédure la moins invasive possible.
- Ils travaillent également aux différentes courbes de libération des principes actifs, comme de l’analgésique, qui pourraient être contenus dans les membranes.
LES PARTENAIRES INSTITUTIONNELS ENTRENT EN JEU
Les partenaires de la Fondation Maladies rares agissent pour l’amélioration de la compréhension et de la prise en charge des personnes atteintes de maladies rares.


M. Étienne VAN DER ELST a présenté accelRare (Sanofi), outil digital de pré-diagnostic différentiel, dispositif médical de classe I, dédié aux maladies rares pour lutter contre l’errance et l’impasse diagnostiques. Cet outil conçu en lien avec les experts des centres labellisés maladies rares renseigne actuellement sur 270 pathologies en français ou en anglais. Il permet aux médecins du réseau primaire de soins, à partir des signes des patients, même face à une situation clinique atypique, de suspecter une possible maladie rare, et d’orienter rapidement vers le bon parcours de soin (centre expert le plus proche, filières de santé maladies rares).
L’outil permet, en moins de 10 minutes :
- D’accompagner le médecin dans sa démarche diagnostique ;
- De fournir la fiche descriptive complète pour mieux se réapproprier la maladie ;
- De proposer une liste d’examens complémentaires à réaliser en libéral pour asseoir le niveau de suspicion de la maladie ;
- D’indiquer la filière au plus proche du patient.
Mme Myriam GEHCHAN – UCB (union chimique belge), entreprise biopharmaceutique à visée internationale, implémentée dans 36 pays avec près de 9000 employés et 3 millions de patients qui ont bénéficié de traitements en 2023, a présenté l’ambition maladies rares.
Son héritage scientifique est solide avec un fort engagement en recherche et développement dans les domaines thérapeutiques principaux des neurosciences et de l’immunologie ce qui lui a permis d’évoluer du domaine de la chimie, vers un groupe pharmaceutique et maintenant la biopharmaceutique spécialisée en épilepsie et maladies auto-immunes.
L’ambition du futur s’ouvre aux maladies rares, thérapies géniques et au développement de l’intelligence artificielle en R&D.
Ses objectifs :
- Améliorer la prise en charge des patients ;
- Contribuer et apporter des solutions qui améliorent leur vie au quotidien.
LES APPROCHES SOCIALES ET HUMAINES POUR AMÉLIORER LA QUALITÉ DE VIE
La matinée s’est terminée sur les approches sociales et humaines qui visent à améliorer la vie des patients atteints de maladies rares. Les recherches exposées portaient sur :

L’efficacité du programme d’ETP dans la myasthénie avec l’étude de preuve de principe (MY-EDUC) qui se focalise sur la qualité de vie des patients souffrant de cette maladie chronique, présenté par le Dr Damien OUDIN DOGLIONI, laboratoire LIP/PC2S – Université Grenoble Alpes – Grenoble, co-lauréat avec le Dr Aurélie Gauchet de la bourse de recherche pour l’amélioration concrète de la qualité de vie des patients atteints de la myasthénie auto‑immune soutenue par le laboratoire UCB.
De l’incertitude des savoirs à l’incertitude de soi dans le syndrome de Turner par Mme Eva LAIACONA, doctorante à l’UMR 7069, laboratoire interdisciplinaire en études culturelles (LinCS) – Université de Strasbourg sous la direction du Pr Nicoletta DIASO dont la bourse de thèse a été financée par la région Grand-Est et Novo Nordisk.

Cette étude porte sur l’expérience et la médicalisation de femmes, jeunes filles, qui sont touchées par le syndrome de Turner (absence totale ou partielle d’un chromosome X) et plus particulièrement sur les traitements hormonaux grâce à des études qualitatives (observations lors de consultation, en réunion associative, entretiens).
À travers les témoignages des patientes, la stigmatisation, « être traitée plus bas que terre » a marqué la plus ancienne génération et même si cette figure archétypale peut demeurer, cela donne à présent des positionnements délicats et ambivalents selon les interlocuteurs tant du côté des familles que des professionnels de santé.
Ce qui ressort des observations, c’est la volonté actuelle de ne pas stigmatiser et on peut avoir tendance à occulter la différence, infantiliser mais cette intention à ne pas normaliser peut avoir comme effet pervers d’invisibiliser certaines difficultés réelles auxquelles font face ces jeunes filles et femmes.
Mme Laetitia CLABAUT & Mme Marjolaine CORBEIL, psychologues au service de génétique clinique, CHU de Lille, ont pu présenter l’étude SURGENEA qui porte sur l’accompagnement des parents confrontés au diagnostic de surdité permanente néonatale d’origine génétique : analyses des difficultés et des besoins.
La surdité permanente néonatale est un déficit sensoriel qui n’est pas rare puisqu’il concerne 1 naissance sur 1000 par contre elle englobe un ensemble de maladies rares dont 60 à 80 % sont d’origine génétique malgré des parents entendants. Le dépistage a été rendu obligatoire depuis le 23 avril 2012 et permet une prise en charge précoce de l’enfant cela étant les parents ignorent souvent tout de ce déficit. C’est un processus d’annonce graduelle en maternité, puis de diagnostic chez l’ORL et enfin de diagnostic génétique.

Cette étude a pour spécificité d’étudier le vécu parental des mères mais aussi des pères de façon isolée avec des entretiens semi-directifs à ces 3 étapes du diagnostic pour pouvoir améliorer la compréhension des difficultés, mettre en exergue les besoins et ajuster l’accompagnement.
48 couples ayant eu un enfant entre 2012 et 2022 ont été inclus. Les résultats qualitatifs montrent que c’est un processus discontinu avec un vécu émotionnel qui va évoluer sur les 3 étapes, et qui sera différent du côté des pères et du côté des mères. Cela questionne sur les informations à communiquer à la maternité, de ne pas sous-estimer les conséquences émotionnelles chez les pères, de favoriser la présence des 2 parents lors de toutes les étapes pour que la mère ne soit pas le messager et d’adapter le soutien.
Mme Michalina DANNOUNE, psychologue clinicienne dans le service de médecine interne des maladies infectieuses du CHU de Versailles, lauréate de la bourse de recherche Novo Nordisk pour l’amélioration de la qualité de vie des patients atteints de drépanocytose a pu présenter l’étude Drépa-FACETS inspirée de FACETS (pour la sclérose en plaques).

Cette étude a été menée avec l’université Grenoble Alpes et l’association SOS-Globi. La drépanocytose concerne 1/1323 naissances en 2020, elle est ainsi un enjeu de santé publique peu étudié sur ses aspects de retentissements psychologiques dues à la douleur (marqueur de sévérité) et la fatigue (93,6 % des patients).
FACETS est un programme de 6 séances de 90 minutes proposées sur 6 semaines pour 6 à 8 participants qui visent à normaliser l’expérience de la fatigue, d’apprendre aux participants à utiliser leur énergie et de développer un mode de pensée utile concernant la fatigue pour la gérer au mieux au quotidien et ainsi améliorer leur qualité de vie.
Cette étude sera menée de septembre 2024 à décembre 2025.
FOCUS POSTER
La pause déjeuner s’est imposée comme un moment suspendu, café à la main après le repas, pour l’étude des 27 posters affichés dans les couloirs du prestigieux campus des Cordeliers.
Nous retiendrons les travaux de Kristelle EL JEKMEK et d’Anaïs SAINT-MARTIN WILLER, doctorantes sous la direction du Pr David MONTANI, du Dr Fabrice ANTIGNY et du Pr Marc HUMBERT.
Leur thèse porte sur le rôle crucial du CRACR2A dans la pathogénèse de l’hypertension artérielle pulmonaire.




